
Aligning AI Use Clinical Trials With FDA And EMA Expectations
By Jessica Cordes, senior consultant for clinical operations, Clinical Excellence GmbH

Even when strategic decisions, vendors, and data science teams sit in the United States, the moment a clinical trial is conducted in EU/EEA countries, European regulators will expect the sponsor to demonstrate that any AI used across clinical trial planning, conduct, and analysis is transparent, controlled, and fit for purpose within the European regulatory context. The recent publication of the EMA’s Reflection Paper on the Use of Artificial Intelligence (AI) in the Medicinal Product Lifecycle1 marks a critical milestone for anyone involved in clinical development, especially U.S.-based sponsors who plan or already conduct clinical trials in Europe.
The reflection paper captures EMA’s current expectations and is a strong indicator of what reviewers may ask for during scientific advice, clinical trial applications, and ultimately marketing authorization discussions. For U.S. companies, this means aligning internal AI practices with EU expectations for GxP, data integrity, transparency, and oversight, and doing so early enough to avoid late-stage questions or clinical trial delays in Europe.
The EMA outlines a human-centric and risk-based approach to AI throughout the medicinal product life cycle. The scope extends from preclinical development to post-authorization monitoring. For clinical operations, the most immediate impact is on clinical trial planning, execution, and regulatory submission.
Any AI application that has a potential impact on patient safety, benefit-risk assessment, or regulatory outcomes must be treated as part of the clinical development dossier. This includes tools used in patient selection, randomization, recruitment, endpoint analysis, and even those used to assist with data cleaning or signal detection.
From a regulatory point of view, the EMA expects full transparency when AI tools are used in clinical trials. This includes access to model architecture, development logs, validation processes, training data summaries, and a complete description of how the AI integrates into the data processing pipeline.
Guiding Principles Of Good AI Practice In Drug Development
In January 2026, the FDA and EMA published Guiding Principles of Good AI Practice in Drug Development,2 a set of 10 high-level principles intended to foster responsible innovations and promote internationally aligned expectations for AI used to generate or analyze evidence across the drug life cycle. This joint document is particularly useful for U.S.-based sponsors operating in Europe because it provides a shared language to align U.S. internal governance with EU regulator expectations and to demonstrate that AI controls are not EMA-only add-ons but part of a coherent global quality approach.
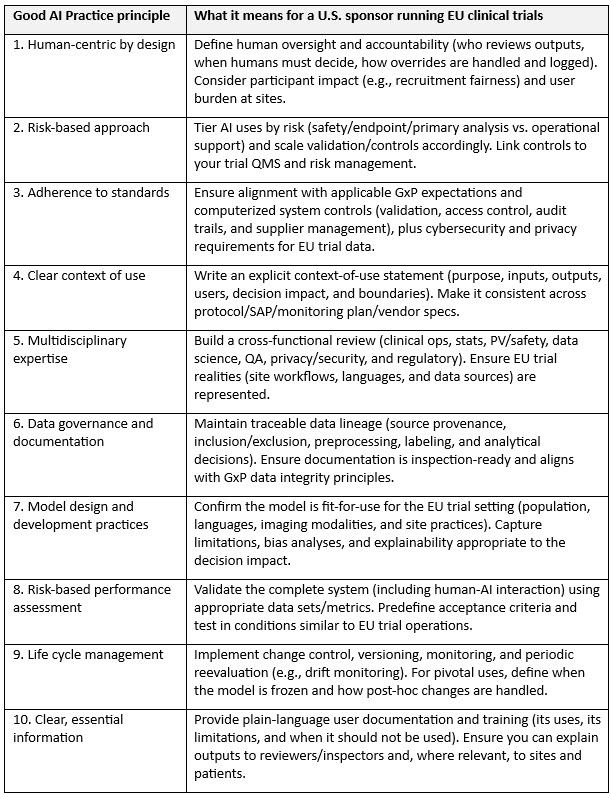
Implications For Clinical Operations
Three themes matter most when running clinical trials in Europe:
- Only the sponsor is responsible, regardless of where the algorithm sits. If an AI system influences clinical trial conduct, data, or conclusions, EU regulators will hold the sponsor accountable even when the model is built by a U.S.-based data science team or an external vendor.
- Regulatory scrutiny extends beyond the EMA. AI use that affects patient safety, endpoint interpretation, or data integrity can trigger questions during clinical trial authorization and inspections, not only at marketing authorization.
- Cross-border data and systems must be governed, not assumed. If AI relies on EU participant data, EHR extracts, imaging, wearable streams, or centralized monitoring outputs, you need clear controls for data quality, access, change management, and (where applicable) international data transfers.
Practical Checklist For U.S. Clinical Teams Running EU Trials:
- Map where AI is used. Report whether it is used for protocol design, feasibility/site selection, recruitment, central monitoring, medical coding support, data cleaning, endpoint assessment, and/or statistical modelling, and label which uses could impact safety, efficacy, or primary analyses.
- Define a clear context of use and decision rights. Report what the model does, what it must not be used for, who uses it, how it influences decisions, who reviews outputs, what happens when humans override it, and how those overrides are logged (plain language where possible).
- Life cycle management (version control + monitoring). Define the model/version used per clinical trial, implement change control, and monitor performance over time (e.g., to detect drift). For pivotal uses, lock the model before confirmatory analyses and document how any updates would be handled.
- Vendor due diligence and contracts. Ensure you can obtain development/validation summaries, training data provenance at an appropriate level, performance metrics and acceptance criteria, known limitations/bias analyses, change logs, user documentation/training materials, and audit/inspection support from an AI vendor, should you use one.
- Prespecify AI use in study documentation. Ensure AI-supported processes are reflected in the protocol, monitoring plan, and SAP where relevant, with clear rationale and risk controls.
- Data protection and cross-border considerations. Confirm how EU participant data flows to U.S. systems (and to any sub-processors), and make sure privacy, security, and access controls are defined and followed.
- Inspection readiness. Maintain traceability from raw data to preprocessing, model output, human decision, and final data set/analysis. Be able to explain how bias was assessed and mitigated.

The immediate implication for clinical operations is the need to treat AI-supported tools with the same level of diligence applied to investigational products and statistical methodologies. This requires clinical trial teams to collaborate closely with data scientists, software vendors, and regulatory colleagues to ensure traceability, compliance, and quality assurance.
The reflection paper urges early engagement with regulators. If an AI application could affect the clinical trial outcome or patient safety, developers should seek scientific advice or qualification procedures early in the process. This advice applies even when the tool is not classified as a medical device. Its influence on data integrity, bias, or endpoint interpretation can make it a regulatory consideration.
Equally important is the documentation of how AI is being used within the protocol and statistical analysis plan. Models must be frozen prior to unblinding, and their use must be prespecified. The EMA explicitly warns against incremental learning or ongoing model optimization during clinical trials, as these practices compromise data integrity and increase the risk of overfitting.
Misconceptions To Avoid
There are several common misconceptions that can hinder compliance. First, not all AI tools need to be certified medical devices to fall under regulatory scrutiny. If a system influences how patients are selected, assigned to treatment arms, or how endpoints are evaluated, regulators may request documentation and validation, even if the system is not CE-marked.
Another frequent misunderstanding is that AI models can be updated or fine-tuned during an ongoing clinical trial. EMA guidance is clear: Models must be frozen before database lock, and any changes after unblinding mean that analysis results will be considered exploratory and not suitable as confirmatory evidence.
Some teams believe that tools used only for recruitment or site selection don’t need to be included in regulatory submissions. This is also inaccurate. If these tools introduce bias, alter patient demographics, or influence clinical trial feasibility, they fall within the scope of the benefit-risk assessment.
Where To Start: Managing AI In Your Clinical Trial Portfolio
The first step is to take inventory of AI-based or data-driven systems currently used or planned for use in your clinical development programs. This includes systems involved in patient identification, protocol design optimization, endpoint analysis, and data review. Once identified, ensure that model documentation is available and accessible. This includes a description of the training data, validation strategy, intended use, performance metrics, and any associated risks.
Next, involve your biostatistics and medical teams in assessing the regulatory relevance of these tools. If the AI impacts clinical trial design or outcomes, it should be integrated into the protocol and statistical analysis plan from the outset. If in doubt, initiate an early dialogue with regulators to clarify expectations.
It is also advisable to define a clear ownership structure internally. Determine who is responsible for AI compliance. Cross-functional collaboration is key, as AI touches multiple parts of the clinical trial life cycle.
Finally, consider whether your team is sufficiently trained to assess, validate, and document AI systems. If not, seek training or external support to build internal capabilities. This may include workshops, checklists, or mock inspections to prepare for regulatory review.
Looking Ahead
AI’s potential to improve efficiency, identify patterns, and support complex decision-making is significant. But the responsibility to use it appropriately lies with clinical professionals and clinical trial sponsors.
Regulators are not trying to block innovation. They’re trying to make sure it’s done safely, transparently, and in the interest of patient protection. As the regulatory framework for AI matures, companies that act early will reduce their risk and gain a strategic advantage.
Clinical trial teams in small biotech and pharma companies are particularly well positioned to lead this change. With the right processes and mindset, AI can support, not complicate, your development goals.
References:
- https://www.ema.europa.eu/en/documents/scientific-guideline/reflection-paper-use-artificial-intelligence-ai-medicinal-product-lifecycle_en.pdf
- https://www.fda.gov/about-fda/artificial-intelligence-drug-development/guiding-principles-good-ai-practice-drug-development
About The Author:
 Jessica Cordes is a clinical operations expert with 15+ years of experience planning, governing, and executing complex clinical trials, with a focus on oncology and cell and gene therapies. She combines operational responsibility and GCP expertise with applied AI transformation in regulated environments. She supports project leads, heads of clinical operations, quality functions, and senior biotech leaders who want to integrate AI in a structured, responsible, and compliant way. Her work follows maturity levels from initial orientation to productive use and audit readiness and includes consulting and training on strategic steering, governance models, and role enablement. Implementation is performed exclusively within her own organization. She works without model development or vendor bias and applies AI only where IT security, data protection, and validation requirements are fulfilled. Since 2023, she has been working as an independent consultant and trainer and provides a GCP refresher course and templates via her Clinical Excellence Training Academy.
Jessica Cordes is a clinical operations expert with 15+ years of experience planning, governing, and executing complex clinical trials, with a focus on oncology and cell and gene therapies. She combines operational responsibility and GCP expertise with applied AI transformation in regulated environments. She supports project leads, heads of clinical operations, quality functions, and senior biotech leaders who want to integrate AI in a structured, responsible, and compliant way. Her work follows maturity levels from initial orientation to productive use and audit readiness and includes consulting and training on strategic steering, governance models, and role enablement. Implementation is performed exclusively within her own organization. She works without model development or vendor bias and applies AI only where IT security, data protection, and validation requirements are fulfilled. Since 2023, she has been working as an independent consultant and trainer and provides a GCP refresher course and templates via her Clinical Excellence Training Academy.