
Avoid Launch Delays By Planning For An FDA-Required REMS
By Melissa Landers, senior principal strategic consulting, Two Labs

Picture this: The FDA accepts a manufacturer's NDA, and the manufacturer plans for its impending launch. But shortly before the anticipated approval, the FDA notifies the manufacturer that a Risk Evaluation and Mitigation Strategy (REMS) program is required to market the product.
REMS are FDA-mandated, post-marketing safety programs that can be required to appropriately balance a drug's benefits and risks to support safe use. These safety programs are most often required during the NDA or biologics license application (BLA) process to address risks identified in development.
If not planned for, this one requirement can set the launch back months, possibly a year, depending on the complexity of the REMS. This reality hits hard; realizing it could have been avoided hits harder.
This unfortunate surprise REMS situation happens more often than it should because REMS is a small niche of the drug safety world. Products needing a REMS represent only 62 of the 20,000+ prescription drug products currently marketed in the U.S., according to the FDA1,2. This means that even among regulatory and drug safety experts, REMS experience and knowledge are limited.
As a result, manufacturers often miss warning signs that a REMS may be needed. In turn, they also don't account for the time and effort necessary to develop, implement, and operate a REMS, which can significantly impact launch timing and patient access.
Signs That You Might Need A REMS
The good news is that while REMS expertise is not common, several warning signs of the need for a REMS are easy to spot because of precedents set by approved REMS programs. While the FDA considers the need for a REMS on a case-by-case basis, these precedents offer directional guidance when considering the likelihood of a REMS.
In some cases, there is a risk theme, such as embryo-fetal toxicity, which is seen across approved REMS in several therapeutic areas. In other cases, the risk tracks to the drug class, as with CAR-T therapies and the risk of cytokine release syndrome (CRS). CRS is a risk at the time of CAR-T administration; risks associated with the administration of other therapies, such as anaphylaxis or loss of consciousness, are also red flags for a potential REMS.
Additional risk themes are listed in Figure 1, which is a compilation of risks seen in the FDA’s documentation of the 62 currently approved REMS. If manufacturers identify these types of risks during the development of their product, they need to seek expert assistance in developing their REMS strategy to avoid downstream challenges.
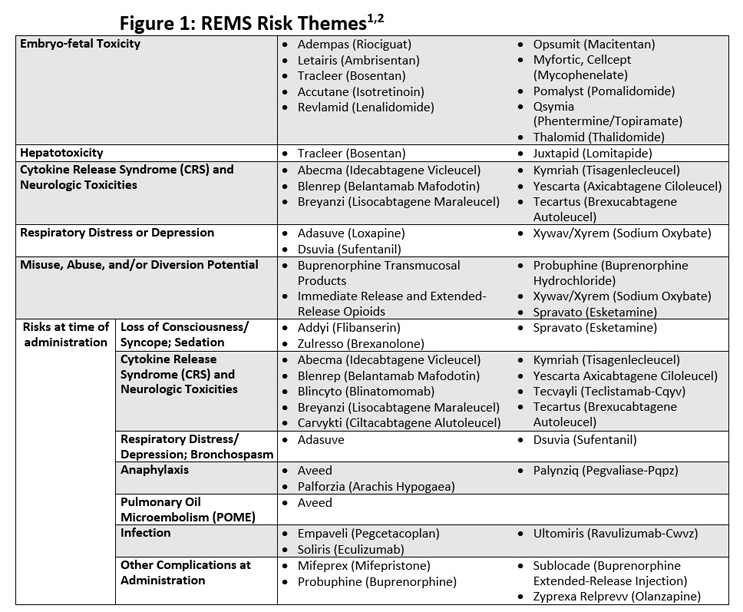
The bad news is that not all risks fall neatly into themes from approved REMS, making them harder to identify during development. FDA considers a drug safe “if the clinical significance and probability of its beneficial effects outweigh the likelihood and medical importance of its harmful or undesirable effects.”2
The challenge is that the FDA applies this definition on a drug-by-drug basis, and the meaning of terms like “likelihood,” “medical importance,” and “harmful or undesirable effects” is largely subjective. So, while manufacturers can find general direction in FDA’s 2019 guidance3 about when a REMS is necessary, consultation with a REMS expert may be needed for more nuanced, product-specific recommendations.
When warning signs for REMS are identified, the first element of the REMS strategy is determining if and when a REMS should be submitted. In the case of risks that fall clearly into common REMS themes, like embryo-fetal toxicity, then it is prudent to engage with FDA about REMS as early as possible and to submit the REMS along with your NDA or BLA. This approach maximizes the time available to design a REMS that supports safe use while minimizing the potential burden on patients, prescribers, and pharmacies.
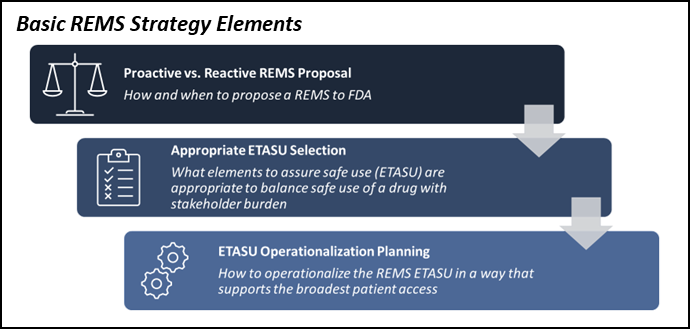
Don’t Be Tempted To ‘Wait And See’
Because REMS are burdensome to stakeholders, manufacturers that find their drug's risk falls in a gray area regarding the need for a REMS often choose to wait and see if the FDA requires one. While this is a solid strategy in this situation, this is also where many manufacturers unwittingly risk launch delays.
During the wait-and-see period, manufacturers should still prepare for the potential FDA REMS request because they will likely not have enough time between the FDA notification and the anticipated launch to write and operationalize a REMS. At a minimum, those who decide to wait and see should write their REMS documentation and identify what would be required to operationalize the REMS. Having the REMS in your back pocket is a form of insurance for the possibility that the FDA asks for a REMS.
A better form of insurance is to also start building the REMS administrator system. REMS require custom systems that typically take six to 12 months to build, and this build time will likely delay your launch if you take a wait-and-see approach with the FDA. While building a custom system that you may not need seems like an unnecessary expense, it is often worth it when weighed against the cost of a launch delay.
Delay to launch is one of many risks associated with waiting until FDA notification to work on your REMS; you also risk patient access due to an unduly burdensome REMS. A hastily assembled safety program is likelier to have burdensome elements and operating procedures because the priority was speed, not quality. When writing the REMS, you must consider the downstream impact of every nuance of each element to ensure safe use.
Plan For The Patient Experience
Seemingly simple REMS design choices, like how often the patient must be counseled on risks, can disproportionately impact patients’ access to the drug. In such a case, prescribers may perceive there is too much burden in having to counsel patients before every prescription refill and, therefore, may choose not to become a REMS-certified prescriber. As a result, this prescriber’s patient would not have access to the REMS drug unless they changed prescribers.
The content of the REMS document must also consider how a REMS administrator system could implement the requirement in our complex healthcare delivery system. Because each REMS is unique, REMS systems are not off-the-shelf technology solutions.
If the REMS system is not constructed thoughtfully, using it can unnecessarily burden stakeholders because it adds additional steps for getting a drug to the patient. For example, if the REMS requires the prescriber to enter labs regularly for their patients, the system should make that process as streamlined as possible. In this instance, simply providing each prescriber with a personal dashboard of their previously accessed patients would save time and effort compared to having to look up each patient anew in the REMs system each month.
When it comes to REMS administrator systems and the contact centers that support them, it is about minimizing patient journey friction. While safe use is the primary objective, you should always remember that reducing the REMS burden that can impact patient access is a close second.
Like other aspects of your launch, your REMS only has one chance to make a good first impression. History has shown that if a prescriber or patient has a negative REMS experience, it is difficult — if not impossible — to get them to try again, even if you improve the program downstream.
Avoid Delays, Be Ready For REMS
In short, if you do not plan for the possibility of a REMS, you put yourself in an impossible situation. Either you lose time to market by taking the time necessary to design, negotiate, and build a high-quality program, or you hinder patient access to your product by hastily assembling a REMS with unnecessarily cumbersome requirements.
While manufacturers may not want to need a REMS, these programs can be thoughtfully planned for, designed, and built to minimize their impact on launch timing and patient access.
Whatever the risk situation for a product, one thing is always true — hoping for the best results does not guarantee the best results. It is always more prudent to engage REMS experts to help you understand and plan for your REMS potential with a REMS strategy.
Note: The list of REMS themes is not exhaustive and is current as of Nov. 3, 2022. For currently approved REMS and their risks, go to https://www.accessdata.fda.gov/scripts/cder/rems/index.cfm.
Resources:
- “Approved Risk Evaluation and Mitigation Strategies.” Food and Drug Administration. https://www.accessdata.fda.gov/scripts/cder/rems/index.cfm. Accessed 11/3/2022.
- "Fact Sheet: FDA at a Glance." https://www.fda.gov/about-fda/fda-basics/fact-sheet-fda-glance
- “REMS: FDA’s Application of Statutory Factors in Determining When a REMS Is Necessary Guidance for Industry.” April 2019. Food and Drug Administration. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/rems-fdas-application-statutory-factors-determining-when-rems-necessary-guidance-industry. Accessed 11/2/2022.
About The Author:
 Melissa Landers has more than 20 years of management consulting experience, with 17 in the biopharma industry. She is focused on supporting regulatory, medical affairs, and commercial launch leadership teams in developing and executing REMS programs, large-scale technology implementations, process improvements, launch strategies, and complex program management. Prior to joining Two Labs, Melissa was a managing director in the Risk and Program Management Advisory Group at Syneos Health Consulting. Melissa holds a bachelors degree in journalism, with an emphasis on marketing and public relations, from the University of Georgia.
Melissa Landers has more than 20 years of management consulting experience, with 17 in the biopharma industry. She is focused on supporting regulatory, medical affairs, and commercial launch leadership teams in developing and executing REMS programs, large-scale technology implementations, process improvements, launch strategies, and complex program management. Prior to joining Two Labs, Melissa was a managing director in the Risk and Program Management Advisory Group at Syneos Health Consulting. Melissa holds a bachelors degree in journalism, with an emphasis on marketing and public relations, from the University of Georgia.