
Beyond The IND Clock: The Legal Architecture Of QRIs And What Industry Must Say Before July 22 (Part 2)
By Kimberly Chew and Odette Hauke
")
Part 1 of this series established that Operation TrialBlazer’s central gap is not the IND review clock but the operational infrastructure between IND readiness and first patient dosed. This article addresses the legal architecture that the pilot leaves unresolved, the compliance asymmetry that its facially neutral qualification criteria obscure, and the specific comments that sponsors and institutions should submit before the July 22 deadline.
Two Anti-Kickback Statute Dockets, Two Distinct Threads
Operation TrialBlazer¹ has surfaced two separate Anti-Kickback Statute (AKS) questions proceeding through two separate comment dockets with different deadlines. They share a statutory foundation but they address distinct relationships, and the comment strategy for each should reflect that distinction.
The Office of Inspector General’s RFI proposed on June 24, with comments due by Aug. 24, 2026, concerns participant-side remuneration: what sponsors may offer clinical trial participants² — transportation, childcare, lost wages, accommodations — to reduce enrollment barriers without triggering AKS liability³ or beneficiary inducement rules.⁴ This is the longer-standing AKS gap in clinical research, and expanding safe harbor coverage here directly addresses Operation TrialBlazer’s most consequential blind spot: enrollment infrastructure. If parallel IRB review and site activation accelerate the IND pathway, but participants still cannot access or afford trial participation, the bottleneck has moved rather than resolved. Comments on the August 24 docket should connect expanded participant remuneration authority explicitly to the enrollment metrics that FDA should build into QRI pilot evaluation.
The FDA’s July 22 RFI surfaces a distinct, institutional-side AKS question. Where a QRI collects fees from a sponsor and then uses its IRB and site infrastructure to support that sponsor’s trial — as the RFI’s own qualification criteria explicitly anticipate — the remuneration-for-referral structure implicates the AKS in a configuration for which no safe harbor was designed. This is categorically different from the participant-remuneration question the OIG’s August 24 RFI addresses. It is especially acute in configurations where a QRI is also the IRB reviewing the trial, the site conducting it, or the employer of the investigators running it. In those arrangements, financial interest, advisory influence, and downstream operational control converge in a single institutional actor. Neither docket currently addresses that convergence.
These two dockets should be treated as analytically linked components of a single regulatory architecture. Comments that engage only one docket in isolation will miss the opportunity to advocate for a coherent framework across both and will leave the institutional-side AKS gap unaddressed.
The Legal Architecture Of QRIs: Fees, Conflicts, And The Limits Of Delegation
The RFI asks whether QRIs should be required to operate their own IRB and/or clinical trial site and separately asks what safeguards would be needed if a QRI serves as both IRB and regulatory advisor. These are the right questions. But the FDA has simultaneously disclosed that QRIs will collect fees from sponsors post-pilot and that the FDA will have no role in setting or collecting those fees. Asking industry to design the conflict of interest safeguards for a fee-bearing intermediary whose fee structure the FDA has explicitly declined to regulate is a significant governance gap, and the July 22 comment window is the appropriate moment to say so.
The legal stakes are concrete. A QRI that (a) collects fees from a sponsor for IND advisory services, (b) operates or is partnered with the IRB reviewing that trial, and (c) operates or is affiliated with the clinical trial sites where the sponsor’s study runs has created precisely the set of financial relationships that AKS analysis is designed to examine. The combination of remuneration, advisory influence, and downstream referral — all flowing from or through the same institutional actor — is the baseline architecture described by the RFI, with built-in conflicts.
Existing AKS safe harbors (personal services arrangements, fair market value, management contracts)⁵ may provide some cover depending on how specific QRI arrangements are structured. But none was designed for a three-way advisory-IRB-site relationship of this kind, and neither the FDA nor the OIG has indicated that existing safe harbor coverage is sufficient. The RFI does not ask this question; the comment record should supply it.
One important ambiguity requires the FDA’s answer: The RFI states that “following the pilot, FDA expects that sponsors will pay fees directly to QRIs” but does not clarify whether QRI compensation is permitted during the pilot itself. Three clarifying questions should be put to FDA: (1) whether QRI compensation will be permitted during the pilot period, and if not, what funding mechanism will support QRI participation; (2) how QRI operating costs during the pilot will be funded or tracked; and (3) how FDA intends to evaluate a pilot whose financial conditions may differ materially from the post-pilot commercial environment. A pilot that demonstrates value when QRI services are provided without charge tells us little about how the ecosystem will function when market-rate fees are introduced.
The delegation question is equally unresolved. The FDA states that QRI recommendations are advisory and that the agency retains full regulatory authority. But if the FDA’s rolling review process treats a QRI-reviewed IND component as materially reducing the agency’s own review burden, questions arise about accountability: What liability does a QRI bear if its recommendation proves materially deficient? What recourse does a sponsor have if a QRI’s conflict of interest screening fails? Can QRI advisory records be subpoenaed in subsequent litigation? The RFI is silent on all of these.
The CRO Advantage: Why Facially Neutral Qualification Criteria Are Not Competitively Neutral
The RFI defines QRIs to include academic medical centers, healthcare networks, CROs, regulatory advisors, and other research organizations, assessed against the same qualification criteria. These categories are not equivalent: the regulatory burden required to function as a QRI differs substantially by institution type, and that differential systematically favors CROs.
An AMC evaluating QRI designation must navigate a compliance landscape that simply does not apply to commercial CROs in the same way. As a Medicare provider, an AMC faces Stark Law physician self-referral analysis.⁶ As a recipient of PHS funding, it is required to maintain and enforce institutional conflict of interest policies,⁷ and a QRI role that generates fee income while the same institution operates the IRB reviewing the sponsor’s trial is precisely the configuration those policies were designed to scrutinize. As a 501(c)(3) nonprofit, an AMC must assess whether QRI fee income constitutes unrelated business taxable income.⁸ As an institution subject to federal cost accounting standards, it must categorize that income consistently with existing indirect cost rate agreements.⁹ None of these constraints apply to a for-profit CRO in the same way.
The comparison across key compliance dimensions is summarized below.
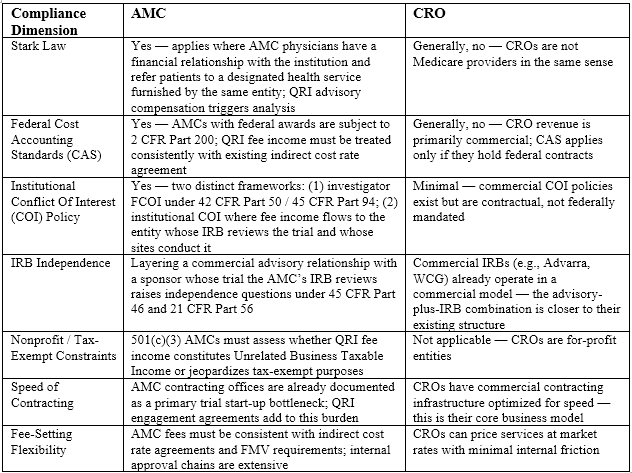
The irony is that the policy narrative driving Operation TrialBlazer specifically invokes “America’s world-class research institutions” — language that signals AMCs. The scientific case for AMC participation is strong: AMCs hold the deepest concentration of early-phase translational expertise in the country, particularly in novel modalities, rare diseases, and first-in-human pharmacology. But if AMC compliance burden and contracting friction make QRI designation impractical for all but the most heavily resourced academic institutions, the program may end up populated primarily by CROs, whose operational speed is real but whose scientific depth in these areas is not equivalent. The FDA should address this asymmetry explicitly, including by providing institution-type-specific compliance guidance for AMC applicants and by building QRI-type composition into the pilot’s evaluation framework from the outset.
What Sponsors Should Say Before July 22
Recommendation 1: Build enrollment and site activation metrics into QRI qualification from day one.
The FDA should require QRI applicants to demonstrate a documented track record on the operational timeline — not only regulatory advisory expertise. Qualification criteria should ask for historical site activation timelines, evidence of participant identification infrastructure, and demonstrated contracting speed. Pilot evaluation should then track time-to-first-patient-dosed, not just time-to-IND-clearance, from the first cohort. Metrics chosen at launch shape behavior; if the pilot measures only submission quality, that is all QRIs will be built to deliver.
Recommendation 2: Ensure QRI capacity is accessible to smaller sponsors and rare disease programs.
If institutions with the strongest advisory and site infrastructure are the most sought after, large pharma will contract with the best of them first, and smaller biotechs and rare disease sponsors risk being crowded out. The FDA should address capacity planning and equitable access explicitly by reserving QRI capacity for small and rare disease sponsors, monitoring access patterns across the first cohort, and treating inequitable access (RFI Question B.1.iii) as a reportable pilot outcome rather than an accepted side effect.
Recommendation 3: Treat the OIG safe harbor RFI as a companion to the QRI pilot.
Sponsors and industry associations should submit coordinated comments on both the July 22 QRI docket and the August 24 OIG AKS docket. The participant-side and institutional-side AKS questions are legally distinct and both deserve coordinated engagement. Comments that treat the two dockets as independent will miss the opportunity to advocate for a coherent framework governing the full QRI ecosystem.
Recommendation 4: Require a robust COI framework before QRIs can serve dual advisory and IRB roles.
The FDA should establish clear rules before the pilot launches on whether a QRI that advises on an IND can also serve as the IRB for that trial. If dual roles are permitted, specific structural safeguards — independent review panels, recusal procedures, audit requirements — should be mandatory. The FDA’s RFI Question A.1.vi asks what safeguards would be needed; the comment record should answer with specificity, not defer to institutional discretion.

Recommendation 5: Define how QRI recommendations interact with FDA’s AI-assisted review infrastructure.
The FDA’s HALO platform consolidates submission data across centers and Elsa 4.0 supports quantitative data analysis during review.¹⁰ Sponsors and QRIs need clarity on how AI-assisted FDA review will interact with rolling QRI recommendations. If the FDA’s AI tools surface concerns that QRIs did not flag, or vice versa, there must be a documented process for reconciliation that preserves the integrity of the administrative record.
Recommendation 6: Define the fee architecture, FMV requirements, AKS safe harbor treatment, and institution-type compliance framework before the pilot launches.
The comment record should ask the FDA and the OIG to address the following before the pilot advances:
- (a) Fee governance — The FDA should clarify whether fees apply during the pilot itself, specify whether QRI fee arrangements must meet a documented FMV standard, identify who sets the FMV methodology, and explain how a fee-free pilot will be a valid proof of concept for a post-pilot commercial environment where market-rate fees apply.
- (b) AKS safe harbor coverage — Comments on the July 22 docket should ask the FDA to coordinate with OIG on whether a new or modified AKS safe harbor is required for the QRI fee relationship, and what structural conditions (documented independence between advisory and site functions, mandatory recusal, audit trails accessible to FDA) would be necessary to qualify.
- (c) Equitable access — The SCRS/Tufts CSDD 2025 Landscape Survey found that 21 percent of clinical trial sites have already declined a trial due to budget disagreements with sponsors;¹¹ introducing a QRI advisory fee with no FMV requirement and no access mechanism adds cost at the sponsor level without correcting the underlying site budget misalignment and will price out pre-revenue rare disease programs before the pilot scales. The pilot should include either a sliding-scale mechanism or a defined fee waiver process, and the evaluation framework should track QRI engagement by sponsor size and therapeutic area from the outset.
- (d) Differentiated compliance guidance — The FDA should provide institution-type-specific guidance acknowledging that AMC and CRO applicants face materially different compliance environments, covering how QRI fee arrangements, COI frameworks, and dual-role structures interact with Stark Law,⁶ federal cost accounting standards,⁹ Public Health Service-mandated institutional COI policies,⁷ and 501(c)(3) unrelated business income analysis.⁸ Treating these institution types as equivalents in the qualification process does not neutralize the compliance asymmetry; it simply leaves AMCs to resolve it internally, at cost and with delay, while CROs proceed unencumbered.
Conclusion
Operation TrialBlazer deserves sincere scrutiny and engagement. The QRI framework, rolling IND review, updated CMC guidance, and dose-selection reforms all target real problems, and each improves on the status quo. But the initiative will close the gap with China and Australia only if QRIs are evaluated on the full pretrial timeline — site activation and enrollment included — rather than on regulatory submission quality alone. The pilot’s fee structure, its silence on the AMC/CRO compliance asymmetry, and its unresolved institutional-side AKS questions are design gaps the comment record can still close.
Two facts sharpen the deadline. The road map and the pilot were both launched under acting FDA leadership, following the commissioner’s May 2026 resignation and with the CBER directorship also unfilled;¹² a program’s design commitments are most open to influence before permanent leadership inherits them and hardest to move afterward. The fee-bearing structure that the RFI describes will turn into commercial practice the moment the pilot ends. July 22 is the last point at which both remain open. Industry should use it.
Process Diagrams
Diagram 1 — Current Clinical Trial Ecosystem (Pre-TrialBlazer)
The current pathway runs sequentially: The sponsor prepares the IND internally (CMC, pharmacology/toxicology, clinical protocol), with an optional pre-IND meeting with the FDA. The formal IND is submitted and the 30-day FDA review clock starts. If a clinical hold is imposed, resolution and resubmission can take weeks to months. Once the IND clears, IRB review begins (local or central, ranging from nine to 35 days per site), followed by site contracting and budget negotiation (two to nine+ months per site), site activation and training, and finally first patient dose. Each step is largely sequential — IRB and contracting cannot meaningfully begin until after IND clearance in the standard model.
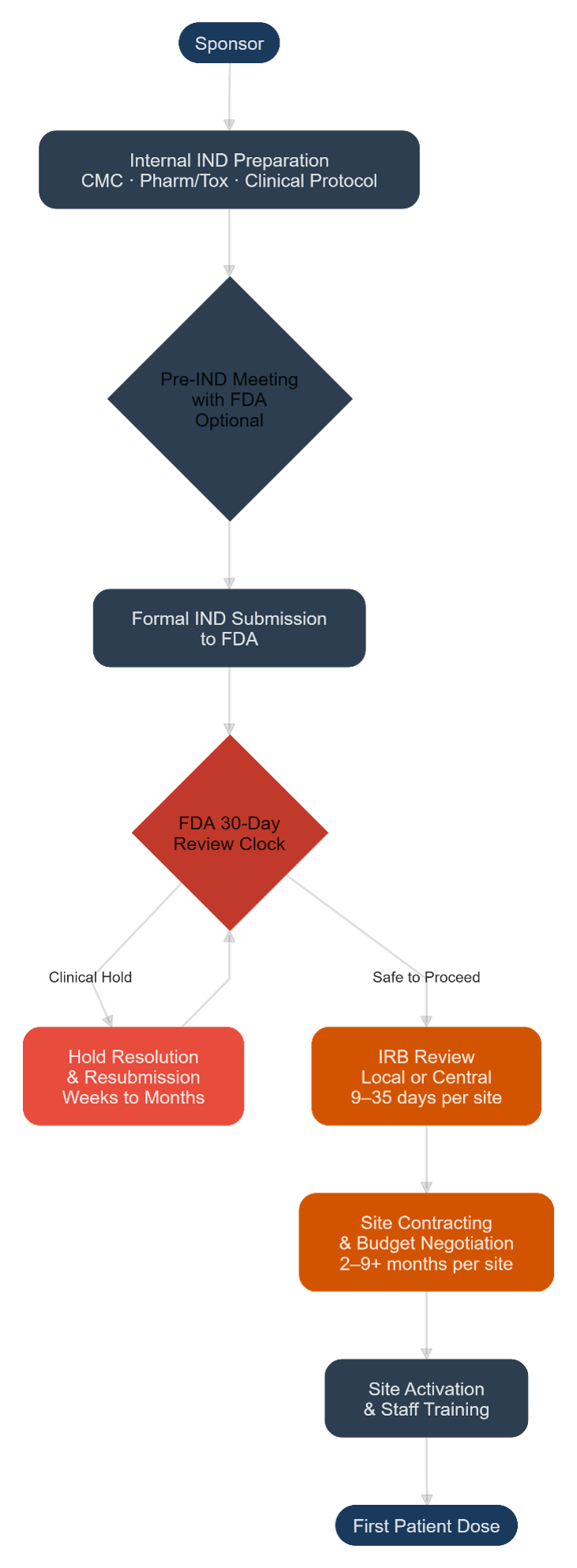
Diagram 2 — Proposed Pilot and Post-Pilot Ecosystem (Operation TrialBlazer)
Under the proposed pilot, the sponsor first engages a QRI under a fee agreement with conflict of interest screening. The QRI conducts a rolling advisory review of IND components (CMC, pharmacology/toxicology, clinical protocol) on an iterative basis, submitting completed components to the FDA via the new rolling platform in real time. Critically, IRB review and site contracting/activation are designed to proceed in parallel with IND development and rolling FDA review — not sequentially after IND clearance. Once the final IND component is submitted, the formal 30-day FDA review clock starts, but because the FDA has already reviewed components on a rolling basis, a safe-to-proceed notification may issue before the 30 days expire. The QRI — which is expected under the qualification criteria to have IRB and clinical trial site infrastructure either directly or through partnerships — is the institutional actor coordinating all of these parallel tracks. Post-pilot, sponsors pay fees directly to QRIs; the FDA sets no rules on fee amounts or FMV.
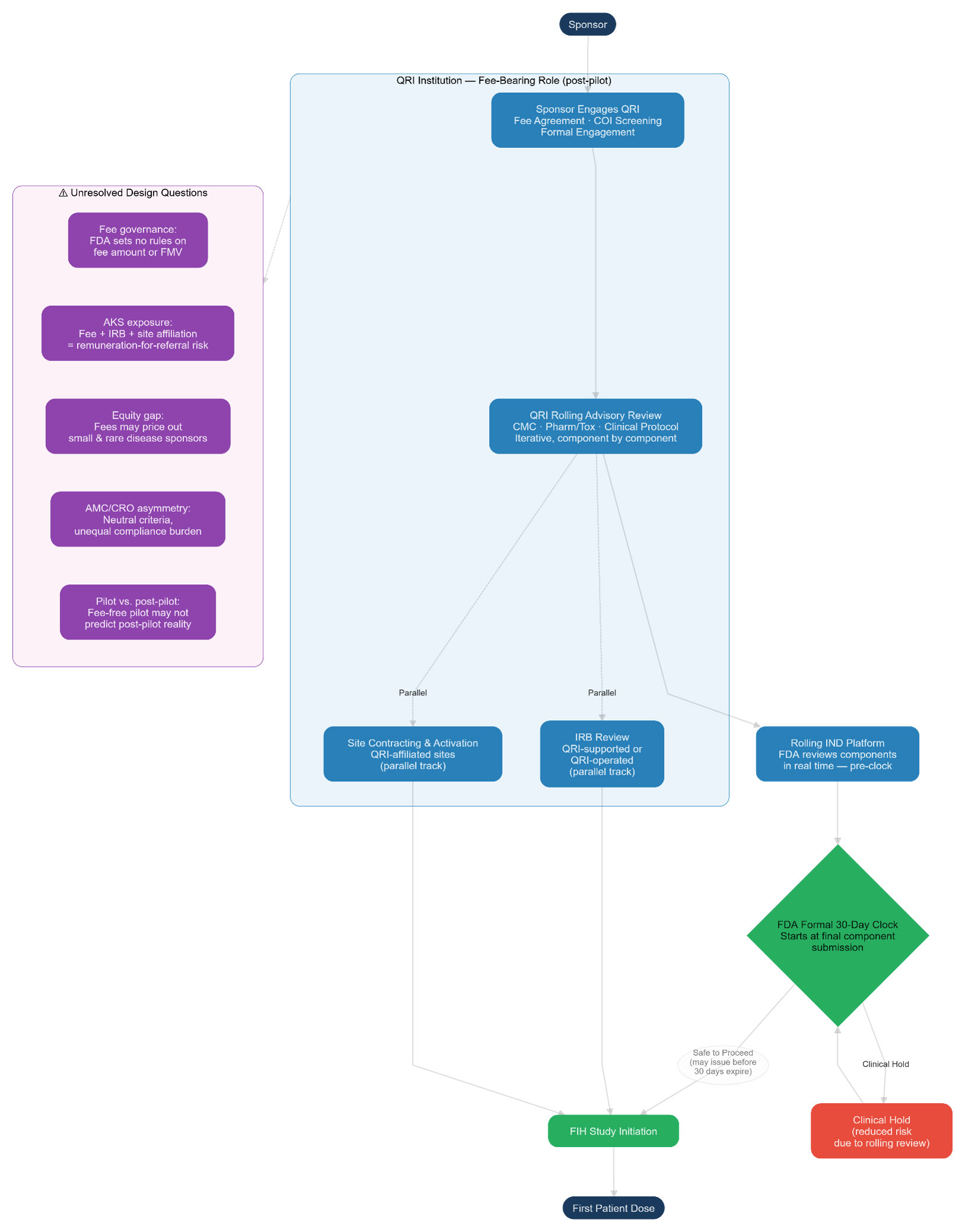
Key unresolved legal questions the pilot does not yet answer: (1) fee governance — the FDA has disclaimed any role in setting or collecting fees, leaving AMCs and CROs to navigate fee-setting without regulatory guidance on FMV or AKS safe harbor coverage; (2) AKS exposure — the combination of QRI fee receipt, IRB operation, and clinical trial site affiliation for the same sponsor’s trial raises remuneration-for-referral questions for which no existing safe harbor was designed; (3) equitable access — fees may disadvantage small and rare disease sponsors; and (4) dual-role COI — the FDA asks but does not answer whether a QRI can serve simultaneously as IND advisor and IRB.
References:
- FDA RFI, U.S. Food & Drug Admin., Request for Information: Expedited Investigational New Drug (IND) Pilot Program, 91 Fed. Reg. 37,996 (June 24, 2026) (Docket No. FDA-2026-N-4699) (Operation TrialBlazer initiative description; Expedited IND Pilot Program generally).
- Medicare and State Health Care Programs: Fraud and Abuse; Request for Information Regarding the Federal Anti-Kickback Statute and Beneficiary Inducements CMP, 91 Fed. Reg. 37,902 (proposed June 24, 2026) (to be codified at 42 C.F.R. pts. 1001, 1003). Comments due Aug. 24, 2026.
- 42 C.F.R. § 1001.952 (Anti-Kickback Statute safe harbors, including personal services and management contracts).
- 42 C.F.R. § 1003.110 (definition of “means of remuneration” under the civil monetary penalty provision for beneficiary inducements).
- 42 C.F.R. § 1001.952 (AKS safe harbors generally).
- 42 U.S.C. § 1395nn (Limitation on certain physician referrals); 42 C.F.R. § 411.350 et seq. (Stark Law regulations).
- 42 C.F.R. § 50 Subpart F (Public Health Service financial-conflict-of-interest regulations for PHS-funded institutions).
- 26 U.S.C. §§ 511–514; 26 C.F.R. § 1.513-1 (unrelated business taxable income).
- Uniform Administrative Requirements, Cost Principles, and Audit Requirements for Federal Awards, 2 C.F.R. Part 200 (§§ 200.414, 200.419); Cost Accounting Standards, 48 C.F.R. ch. 99.
- U.S. Food & Drug Admin., FDA Expands AI Capabilities and Completes Data Platform Consolidation, News Release (May 6, 2026), https://www.fda.gov/news-events/press-announcements/fda-expands-ai-capabilities-and-completes-data-platform-consolidation.
- Jimmy Bechtel & Ken Getz, Old Habits, Communication Issues Still Stalling Site Budget Negotiations, Clinical Leader (March 19, 2026), https://www.clinicalleader.com/doc/old-habits-communication-issues-still-stalling-site-budget-negotiations-0001.
- Marty Makary resigned as FDA Commissioner on May 12, 2026, and Kyle Diamantas was named acting commissioner. Vinay Prasad departed as director of CBER at the end of April 2026. Both the road map (June 22) and the RFI (June 24) issued under acting leadership.
About The Authors:
 Kimberly Chew is co-lead of Husch Blackwell’s Psychedelic and Emerging Therapies practice group. She guides clients through every stage of development, from clinical trial agreements and research collaborations to regulatory compliance and enforcement matters. She represents drug developers, biotechnology companies, and startups, often helping new ventures address early-stage challenges such as corporate formation, trademarks, and governance by collaborating with firm colleagues to deliver integrated legal solutions tailored to each client’s goals and stage of development. She may be reached at Kimberly.Chew@huschblackwell.com.
Kimberly Chew is co-lead of Husch Blackwell’s Psychedelic and Emerging Therapies practice group. She guides clients through every stage of development, from clinical trial agreements and research collaborations to regulatory compliance and enforcement matters. She represents drug developers, biotechnology companies, and startups, often helping new ventures address early-stage challenges such as corporate formation, trademarks, and governance by collaborating with firm colleagues to deliver integrated legal solutions tailored to each client’s goals and stage of development. She may be reached at Kimberly.Chew@huschblackwell.com.
 Odette Hauke is a regulatory scientist and drug development advisor specializing in psychedelic, controlled-substance, and first-in-class therapeutics for serious conditions with significant unmet medical need, with a focus on neuropsychiatry, rare disease, and women’s, maternal, and pediatric health. She is the founder of Regulatory Atelier, a regulatory strategy consultancy operated through Odette Alina, LLC. Across 13 years of global regulatory affairs practice at AtaiBeckley, Vera Therapeutics, Travere Therapeutics, Regeneron, and Memorial Sloan Kettering Cancer Center, she has led regulatory strategy for psychedelic IND programs involving R-MDMA, DMT, and ibogaine. Her expertise spans IND and BLA/NDA strategy, regulatory intelligence, controlled-substance development, AI-enabled regulatory analysis, and direct engagement with the FDA, DEA, EMA, MHRA, and Health Canada. She holds a M.S. in regulatory science from Northeastern University, a certificate in psychedelic science and medicine from Johns Hopkins University, and a B.S. in epidemiology from the CUNY Baccalaureate for Unique and Interdisciplinary Studies.
Odette Hauke is a regulatory scientist and drug development advisor specializing in psychedelic, controlled-substance, and first-in-class therapeutics for serious conditions with significant unmet medical need, with a focus on neuropsychiatry, rare disease, and women’s, maternal, and pediatric health. She is the founder of Regulatory Atelier, a regulatory strategy consultancy operated through Odette Alina, LLC. Across 13 years of global regulatory affairs practice at AtaiBeckley, Vera Therapeutics, Travere Therapeutics, Regeneron, and Memorial Sloan Kettering Cancer Center, she has led regulatory strategy for psychedelic IND programs involving R-MDMA, DMT, and ibogaine. Her expertise spans IND and BLA/NDA strategy, regulatory intelligence, controlled-substance development, AI-enabled regulatory analysis, and direct engagement with the FDA, DEA, EMA, MHRA, and Health Canada. She holds a M.S. in regulatory science from Northeastern University, a certificate in psychedelic science and medicine from Johns Hopkins University, and a B.S. in epidemiology from the CUNY Baccalaureate for Unique and Interdisciplinary Studies.