
Combination Products: U.S. Vs. EU Requirements And A Harmonized Strategy To Prepare CTD Module 3
By Marine Joly-Battaglini, PharmDev Consulting

Recent years have been marked by a high increase in the number of marketed injectable products due to the strong development of biological and biotechnological products. This led to an important expansion of combination products to facilitate administration of the products to patients.
Guidances to help build the marketing authorization dossiers have existed in both the U.S. and the EU (our regions of interests for this article) for some years now and are globally harmonized in terms of the expected content of the information submitted. However, differences exist in the terminology used, as well as in the format and location of the information in the dossiers due to different marketing authorization pathways.
The scope of this article is on only integral products, according to the European Medicines Agency (EMA) definition, or a combination product with a primary mode of action induced by a drug or a biologic, according to the FDA. I’ll detail the regulatory factors involved for both regions and the main guidelines as well as the terminology. I’ll also discuss the main differences between the American and European approaches and I’ll propose a strategy to circumvent these differences.
What Is The Regulatory Landscape For Combination Products In the U.S. And EU?
Who Regulates Combination Products?
This is probably one of the main differences between the U.S. and EU. While in the U.S. a combination product is fully regulated by the FDA (with the support of different centers), in Europe two different and independent entities are involved. The main actors for both regions are described in Table 1.
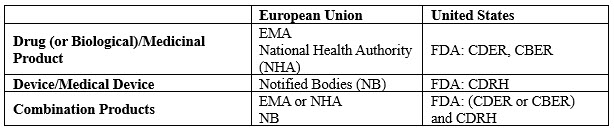
Table 1: The Main Actors in the U.S. and EU Regulating Combination Products
While in the U.S. everything lies with the FDA, and one submission is needed, in the EU, two completely separate entities (a notified body and the EMA or a National Health Authority) review different parts of the dossier, which leads to a submission in two steps.
What are the Main Regulatory Texts to Consider?
In the U.S., two main guidelines are important:
- FDA’s eCTD Technical Conformance Guide2
- FDA’s guidance for industry, Current Good Manufacturing Practice Requirements for Combination Products1
In addition to these guidelines, 21 CFR3 Parts 3, 4, 211, and 820 should be considered.
In the EU, there is mainly one guideline (Guideline on quality documentation for medicinal products when used with a medical device4) that could be seen as an equivalent of the FDA’s eCTD Technical Conformance Guide.2 Additionally, keep in mind the Medical Device Regulation6 and the Directive 2001/83/EC,7 as well as the Questions & Answers for applicants, marketing authorization holders of medicinal products, and notified bodies with respect to the implementation of the Medical Device and In Vitro Diagnostic Medical Device Regulations ((EU) 2017/745 and (EU) 2017/746).5
How Do The FDA And EMA Define a Combination Product?
The definition proposed by FDA in the guideline on cGMP requirements for combination products1 is based on 21 CFR Part 3 and is as follows: “a product composed of two or more different types of medical products (i.e., a combination of a drug, device, and/or a biological product with one another).”
The EMA defines in the guideline on combination products4 a “Drug-Device Combination Product (DDC) as a medicinal product with integral and/or non-integral medical device/device component(s) necessary for administration, correct dosing, or use of the medicinal product.”
In the FDA guidelines, the terminology used includes all types of combination products whereas the EMA guideline defines different types of combination products but always with a primary mode of action as a drug. From the definitions of a combination product proposed by the FDA and the EMA, it seems that the definition used by FDA is broader than the one presented by EMA.
Module 3 Differences
Another difference that can be highlighted is the recommendation made in terms of location of the information in Module 3, which is summarized in Table 2 below (click on image to enlarge).
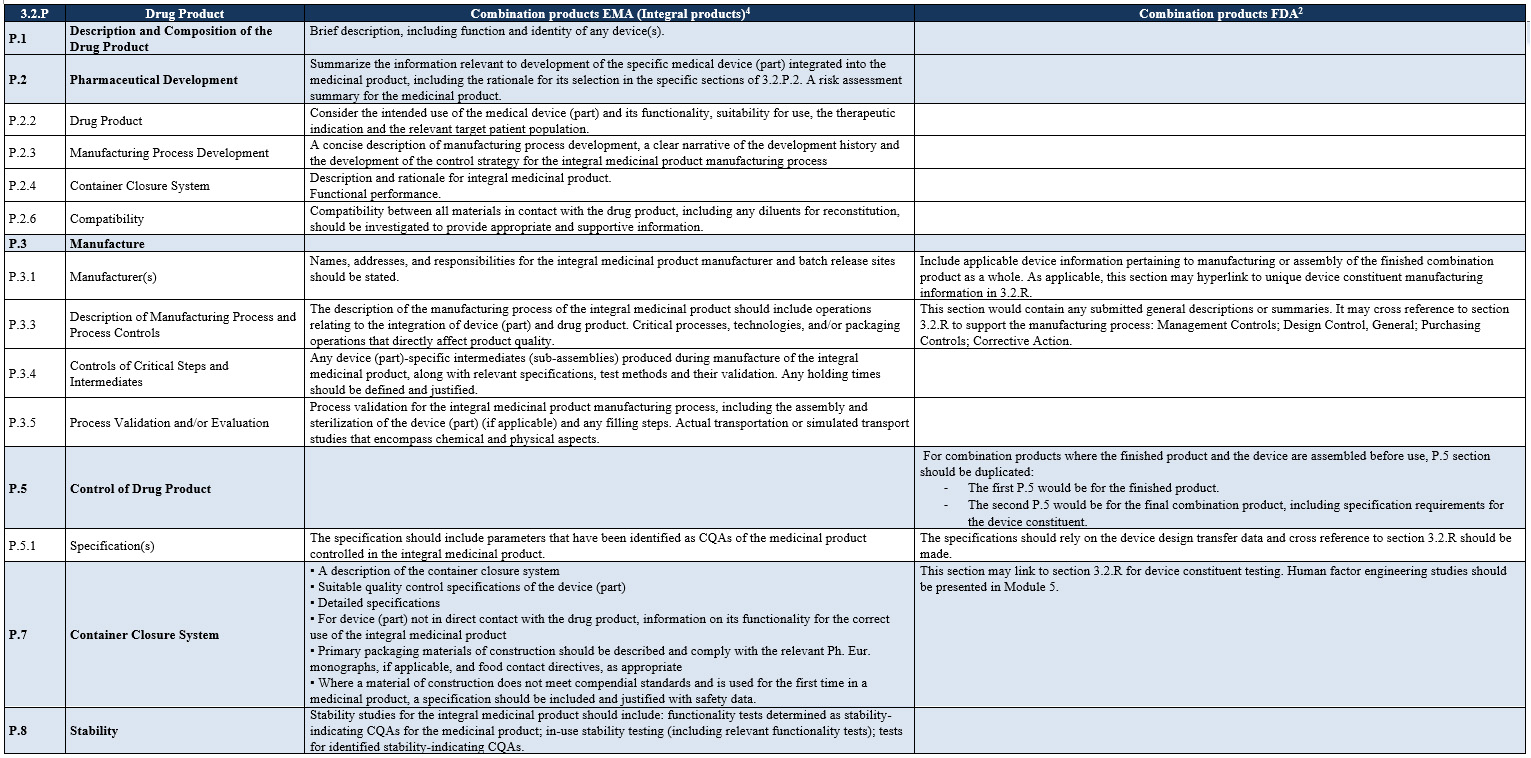
Table 2: Differences Between U.S. and EU in Terms of Location of the Information in Module 3.
Click on image to enlarge.
Given the differences in terms of the location of the information recommended by the FDA and the EMA, it seems that two different Module 3s will need to be prepared: one for each region. This means more time and resources need to be allocated not only for the preparation of the initial submission but also for post-marketing activities, as two dossiers will need to be managed.
Is there not a way to harmonize and streamline the submission preparation when both the U.S. and EU are involved?
Can A Common Quality Module 3 Be Created For Both Regions?
Although the European and American approaches are not completely harmonized regarding the pathway to review and how to present the data within Module 3, the FDA and the EMA are aligned in terms of the expected content (i.e., the studies to be performed and the data to be generated). Therefore, I believe that one Module 3 can be prepared and submitted to both the U.S. and EU.
I recommend using the FDA’s eCTD Technical Conformance Guide2 as a reference for the location of the information and as recommended by this guideline, including most of the information linked to the device Section 3.2.R by creating a specific 3.2.R Device section.
Furthermore, to help the reviewer during the assessment of the dossier, I recommend making cross-references to Section 3.2.R Device in all Module 3 sections where it is normally expected to have information linked to the device, as summarized in Table 3.

Table 3: Proposed Harmonization Strategy
In addition, I recommend preparing a reviewer’s guide (as the FDA recommends), to be submitted in Module 1, that also will help the reviewer quickly locate the most important information.
By adopting this strategy of one dossier based on the FDA guideline, the only adaptation that will need to be performed for the EU is the addition of the Notified Body Opinion (as a stand-alone document) due to the region’s two-step review process.
Conclusion
The landscape for regulation of combination product differs between the U.S. and EU, but with the expected information being the same, these differences can be overcome. It is therefore possible to have a harmonized dossier that can be submitted in both regions by including the requested information in Section 3.2.R of Module 3. When using this strategy, it is important to add the proper cross-references to Section 3.2.R in the appropriate Module 3 sections as well as in the reviewer’s guide. Having most of the information in Section 3.2.R avoids reworking the dossier for an EU or U.S. submission, allows more flexibility for submissions outside of these regions where the product will not necessarily be regulated as a combination product, and facilitates post-marketing activities by limiting the number of sections that need to be updated.
References
- FDA Current Good Manufacturing Practice Requirements for Combination Products
- FDA eCTD Technical Conformance Guide. Technical Specifications Document
- 21 CFR Parts 3, 4, 211, and 820
- EMA Guideline on quality documentation for medicinal products when used with a medical device (EMA/CHMP/QWP/BWP/259165/2019)
- Questions & Answers for applicants, marketing authorisation holders of medicinal products and notified bodies with respect to the implementation of the Medical Devices and In Vitro Diagnostic Medical Devices Regulations ((EU) 2017/745 and (EU) 2017/746)
- EU Medical Device Regulation 2017/745
- EU Directive 2001/83/EC
About The Author:
 Marine Joly-Battaglini is a CMC expert (API and product development) at PharmDev Consulting. She has more than 10 years of experience in the pharmaceutical industry, particularly in CMC development. She has worked in an international environment on a wide range of projects (small and large molecules), at different stages of development (from Phase 1 to the preparation of marketing authorizations, developed in-house or outsourced, including projects that have involved CDMOs and CROs).
Marine Joly-Battaglini is a CMC expert (API and product development) at PharmDev Consulting. She has more than 10 years of experience in the pharmaceutical industry, particularly in CMC development. She has worked in an international environment on a wide range of projects (small and large molecules), at different stages of development (from Phase 1 to the preparation of marketing authorizations, developed in-house or outsourced, including projects that have involved CDMOs and CROs).