
Decentralized Clinical Trials: Does Your Strategy Include These Facets?
By Michael Cooper, Clinical and Regulatory Affairs, Pharmatech Associates

One positive change to come out of the global pandemic is the rapid adoption of decentralized clinical trials (DCTs). In the early days of lockdown, only trial participants suffering from terminal diseases had any incentive to continue participation – at the risk of exposure to a potentially fatal virus at the trial site. Clinical trial enrollment for indications other than oncology plummeted. Since the primary responsibility of sponsors and investigators is to ensure patient safety, it was necessary to move as many trial aspects as possible to the patient’s side. This shift eased travel, cost, and time burdens for patients, allowing trials to maintain their enrollment levels. It also revealed a steep learning curve for sponsors and investigators to deploy mobile healthcare professionals (HCPs) and remote technologies across different states to maintain trial integrity.
Decentralized clinical trials leverage remote access technology to conduct clinical trial activities as close to the patient as possible, rather than at a study site. DCTs have increased in response to the COVID-19 public health emergency, yet this may be due to a convergence of two factors already in play: our reliance on the use of digital health technologies and a move by the FDA and industry toward patient-focused, real-world clinical trials that leverage the technological advances already available.
Basic Elements: Why Decentralization Works
The informed consent (IC) process is itself an example of how activities can be moved closer to the patient. An investigator educates trial participants on the IC process, verifies understanding, and obtains consent. These activities can easily be performed using remote technologies.
Contract research organizations (CROs) have boosted their patient recruitment rates for decentralized trials, thanks to the lowering of time and monetary burdens to participate and increased their savvy for reaching target populations via social media.
Sponsor monitoring of trial sites can easily be performed remotely with existing technologies. In an era of limited travel budgets, a number of the world’s health authorities already perform GMP surveillance inspections simply by requesting documents and asking questions via email. No travel required.
Even the administration of investigational product (IP) can be performed closer to the patient. In a best-case scenario, state law allows shipping oral solid dosage IP directly to the patient for self-administration. More complicated routes of administration require a mobile healthcare professional or travel by the patient to a local medical center. In all scenarios, the sponsor must ensure that proper shipping conditions are maintained and all inventories are reconciled.
Patient assessment for efficacy and safety has benefited the most from technological advances. Implanted or wearable devices collect data continuously, rather than at discrete time points, and transmit data back to the investigator. A mobile HCP may be required for a blood draw, and travel may be required to a local medical center for more sophisticated diagnostic procedures. Figure 1 compares these elements:
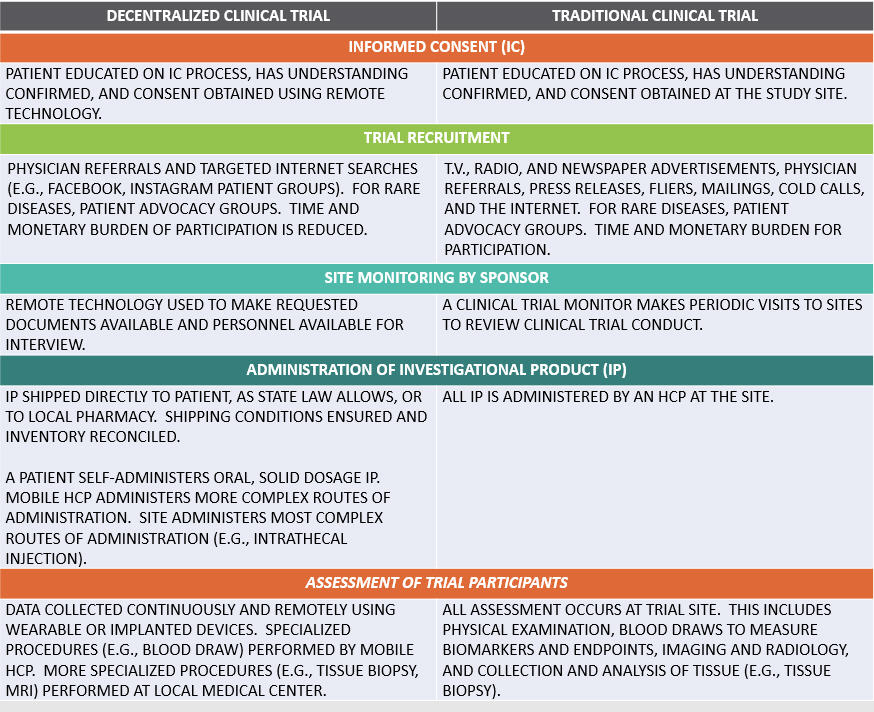
Figure 1 – DCTs vs. Traditional Clinical Trials
Digital Health Technologies Win Use
The Apple Watch is a wearable device we are all familiar with, and millions of consumers worldwide rely on it to monitor their heart rate and heart rate variability at rest, during exercise, sleep, and even meditation. Although inappropriate for use in a clinical setting (it has not been qualified for use and does not undergo routine calibration), it illustrates how far technology has come.
A different type of implanted device, developed for clinical use, can monitor and record heart rhythm data, filtering out atypical rhythms to identify arrhythmia more quickly. Similarly, there is now a device that allows a doctor to remotely measure heart rate and O2 levels and examine eyes, ears, and throat.
Digital technologies are widespread: I volunteered for a mobile adverse event reporting system from the Centers for Disease Control and Prevention (CDC) called V-safe when receiving my two COVID-19 vaccine shots. Periodic text messages guided me to an app to report adverse reactions in a multiple-choice questionnaire.
FDA And Industry Efforts
The FDA has long championed initiatives to make clinical trials more diverse, patient-friendly, representative of the general population, and based on healthcare as delivered in the real world.
Patient-Focused Drug Development Initiative (PFDD)
The idea is to incorporate the patient’s perspective into the design — determining primary and secondary endpoints — and conduct of clinical trials. The FDA takes into consideration the patient’s perspective when reviewing new drug applications (NDAs) and biologic license applications (BLAs).
Traditional measures of a drug’s efficacy (clinical outcome assessments) are performance outcomes (e.g., measuring how far a subject can walk in 6 minutes), and clinician-reported outcomes (i.e., a trained professional’s assessment of a subject’s response to treatment). Under the PFDD model, more weight is placed on patient-reported outcomes and observer-reported outcomes (i.e., a parent or caregiver’s perspective).
A 2017 report observed that Patient Experience Data Tables are included in 82% of NDA and BLA reviews for new molecular entities, indicating that the FDA is incorporating PFDD into the majority of its application reviews. Further assessments will be published by the FDA later this year, in 2026, and in 2031.
Real-World Evidence Program (RWE)
This program’s goal is to incorporate data routinely collected about a patient into the drug development and approval process. Data may come from doctors’ visits, medical information submitted to insurance companies, and product/disease registries.
The randomized control trial has long been the standard for demonstrating a drug’s efficacy. Patients with co-morbidities are often excluded so that the drug’s effect can be more clearly assessed. However, this creates a divergence between the population for whom the drug is intended and the population testing the drug during the clinical trial. Another variance is that in a trial, administration of the IP takes place at a centralized site, whereas in the real-world evidence program, most drugs are administered in a doctor’s office, or self-administered.
Real-world evidence has been in use since the agency launched the Sentinel Program to track post-market surveillance of approved products, in 2008. But rare disease developers have long used natural history studies of patients without treatment as a control arm in their clinical trials, where withholding IP would be unethical. (This approach allowed for the approval of rare disease drugs Myozyme and Brineura.)
Digital Health Information Action (DHIA) Plan
This plan was issued in July 2017 by the FDA’s Center for Devices and Radiological Health (CDRH). The intent is to ensure that all patients benefit from advances in digital technologies, that guidance documents for industry are updated appropriately, and that a Digital Health Software Precertification Pilot Program (Pre-Cert) is developed.
Pre-Cert is the most significant of the DHIA plan’s goals. Traditionally, medical device oversight was focused on hardware. With advances in digital health technologies, software is becoming a more significant area of oversight. The regulatory paradigm needs to be updated because software goes through more iterations during design, development, and validation than hardware does. The Pre-Cert program intends to replace the premarket submission program of digital health technologies with a program for developers of software-based medical devices. Developers certified to have an organizational culture of quality and excellence receive a more streamlined review of their products.
Clinical Trials Transformation Initiative (CTTI)
The FDA and Duke University formed CTTI in 2007 to improve the quality and efficiency of clinical trials. CTTI’s DCT Project generated recommendations on possible protocol designs, navigating state licensing issues around telemedicine and shipping IP, and whether investigators need to be listed on FDA Form 1572.
The Decentralized Trials and Research Alliance (DTRA)
DTRA is a similar public-private partnership but was formed in December of 2020, well into the pandemic. It is focused on some of the more “nuts and bolts” aspects of DCTs, such as nomenclature and definitions, archetypes, and KPIs. DTRA also identifies, documents, and promotes best practices within its membership.
Paying Attention To Real-World Data
One challenge of using real-world data is that a larger effect from the intervention must be demonstrated than in a randomized control trial.
Secondly, such studies use electronic data capture (EDC) systems to collect information from electronic health records (EHRs). Healthcare systems typically manage electronic health records, and they are not Part 11-compliant (e.g., restricting user access, audit trail documenting changes, etc.). Sponsors have to navigate multiple EHRs that may not be interoperable with each other. In all these instances, manual transcription introduces the possibility of transcription error.
Drug sponsors have adopted strategies in response to these concerns, such as creating an external scientific advisory panel or partnering with a data science vendor that specializes in combining data sets from different sources. By including previously published academic scientists and physicians, the sponsor ensures that data will be of higher quality, publishable in a peer-reviewed journal, and able to withstand agency scrutiny.
A third challenge in managing decentralized clinical trial data is assuring the privacy of a patient’s protected health information (PHI), as required by HIPAA and GDPR regulations, while being able to link data to a patient.
One solution is to create a unique identifier with only general information (e.g., gender, region of the country) while excluding protected information. Another solution is to use data tokenization, where sensitive data is replaced with non-sensitive substitutes without altering the type or length of data. Only the tokenization system is able to convert the non-sensitive substitute information back to its sensitive form, reducing the risk of PHI being released inadvertently or by hacking.
The FDA envisions a future state where all electronic health record information is in the form of structured data, allowing it to be extracted by the electronic data capture system. Sponsors will still need to perform validation to demonstrate true interoperability.
COVID-19 Guidance
The FDA issued Conduct of Clinical Trials of Medical Products During the COVID-19 Public Health Emergency in March 2020, updated in January 2021. This is guidance for sponsors, investigators, and Institutional Review Boards (IRBs) on how to continue clinical trials during the pandemic through adoption of DCTs.
Trial participant safety should drive all decision-making and sponsors must weigh the risk of discontinuing treatment vs. the risk of contracting COVID-19 for the patient. Should a patient discontinue treatment, the sponsor needs to assess whether additional safety monitoring is required.
Acknowledging that a shift to remote assessments mid-trial would generate a large number of protocol modifications, deviations, and amendments, the FDA warned about introducing uncertainty to data. If an in-person assessment is switched to a remote technology (e.g., video), then investigators should be consistent in using that same technology for future assessments (i.e., conduct all future assessments by video).
Conclusion: Equal Opportunity?
Historically, patients with terminal diseases and sufficient means could move to a medical center conducting clinical trials (e.g., The University of Texas MD Anderson Cancer Center, etc.) to extend their lives. With decentralized trials, that is now possible for terminal patients with limited means. This levels the playing field for patients and broadens the data collected to a larger diversity of patients across ethnic, racial, and socioeconomic lines, to the benefit of the sponsor, investigator, and overall understanding of the drug.
Survey data gathered by the CRO WCG Clinical Services in April 2021 indicates that most trial sites will continue decentralized trials past the pandemic. Nearly three-quarters (73%) of sites plan to use remote coordinators (vs. 12% prior to the pandemic), 61% plan to use telemedicine (vs. 15% prior), and 93% plan to use remote monitoring (vs. 74% prior). Analysis by the health news website Stat from August 2021 paints a more complicated picture. After peaking in May 2020, telemedicine use has decreased by 60%, with the decline attributed to reimbursement gaps. Revisions are in order for both government and private healthcare plans to continue equitable drug development.
References
- “Assessment of the Use of Patient Experience Data in Regulatory Decision-Making, Final Report,” FDA and Eastern Research Group, Inc., 18 June 2021.
- Webinar: “How RWE is Transforming Clinical Trials,” Clinical Leader Live, 03 August 2021.
About The Author:
 Michael Cooper is a clinical and regulatory affairs program manager at Pharmatech Associates. He has over 20 years of experience in the biopharmaceutical industry, with expertise in regulatory affairs chemistry, manufacturing, and controls (CMC) submissions; GMP inspections for biologics and vaccines; QA lot release of drug substance and drug product; deviation and CAPA resolution; and facilities, utilities, and equipment validation.
Michael Cooper is a clinical and regulatory affairs program manager at Pharmatech Associates. He has over 20 years of experience in the biopharmaceutical industry, with expertise in regulatory affairs chemistry, manufacturing, and controls (CMC) submissions; GMP inspections for biologics and vaccines; QA lot release of drug substance and drug product; deviation and CAPA resolution; and facilities, utilities, and equipment validation.