
FDA Seeks Comment On Design And Data Considerations For Externally Controlled Trials For Drugs, Biologics
By Mark Durivage, Quality Systems Compliance LLC

On February 1, 2023, the FDA released for public comment Considerations for the Design and Conduct of Externally Controlled Trials for Drug and Biological Products. This guidance provides recommendations to sponsors and investigators, addresses considerations for the design and analysis of externally controlled trials, describes considerations related to communicating with FDA, and ensures access by the FDA to data from an externally controlled trial. This guidance does not address the applicability of real-world data or using external control data to distinguish the effects of the medical product from those caused by other factors in a traditional randomized controlled clinical trial or the use of summary-level estimates instead of patient-level data.
When study populations in externally controlled trials are not randomized, the study populations must be as similar as possible to give the best chance of accurately measuring outcomes. Sponsors and investigators, prior to conducting externally controlled clinical trials, need to ensure the trial can differentiate the drug effect from other factors and meet regulatory requirements.
Design Considerations
Applying the correct design elements increases the confidence in study results when appropriate analytic methods are utilized to estimate treatment effects, reducing the bias in externally controlled trials.
The study protocol should be finalized by the sponsor before initiating the externally controlled trial, including selection of the external control arm and analytic approach. The study protocol should specify suitable study data sources, inclusion and exclusion eligibility criteria, exposure definitions and windows, defined endpoints, cogent analytic plans, and approaches to minimize missing data. The study protocol should consider systematic errors in the design, conduct, analysis, or interpretation resulting in an erroneous estimate of a treatment’s effect on the outcome of interest, distortion of the measure of the effect of a treatment on an outcome due to another factor that is associated with both the treatment and the outcome, and events occurring after treatment initiation that affect either the interpretation or the existence of the measurements associated with the clinical question of interest.
When an externally controlled study is not randomized, patients’ attributes that are likely to influence outcomes in an external control arm will differ from corresponding attributes of participants in a treatment arm of the trial due to dissimilar demographic and related factors including age, sex, race, socioeconomic status, and geographic region.
Specific challenges for baseline characteristics can include whether confounding factors are known and well characterized, whether such factors are captured, whether these factors have been assessed with appropriate methods and measured similarly across compared groups, and whether the study’s analytic methods sufficiently address the differences in clinical characteristics between the compared groups.
Externally controlled trial protocols should include plans for evaluating eligibility criteria to determine if the criteria can be applied in a manner that allows for selection of similar patients in the treatment and external control groups. Clinical trials that are properly designed and randomized can generally distinguish between the investigational drug’s efficacy and safety. However, externally controlled trials may not be able to distinguish between the investigational drug’s efficacy and safety due to concerns over the control arm that are either not documented or cannot be accounted for.
Collecting data on the use of concomitant or supportive therapies is generally included in clinical trial protocols, including drug formulation, dose, strength, route, timing, frequency and duration, rules for dose modifications, interruptions, or discontinuations.
The treatment and delivery of care that patients receive can influence the assessment of outcomes related to those treatments, including differences in health-seeking behaviors, insurance coverage, adoption of clinical practice guidelines, availability of novel treatments, use of companion diagnostic testing, and access to emergency department or intensive care.
Specifying the start (index date) of the observation period for assessing endpoints is a challenge with externally controlled trials. Differences in determining the index date across externally controlled trial arms may lead to bias in the estimates of effects. Randomized trials usually designate the index date for the treatment and control as the time when eligibility criteria are determined to have been met and a decision was made regarding the intended treatment strategy for each participant. To avoid bias, determination of the index date in the treatment arm and the external control arm should avoid the period of time during which the outcome of interest could not have occurred in one of the two arms.
Knowledge of the treatment by patients, caregivers, clinicians, or investigators may lead to a biased estimate of the effect of treatment when the trial is not blinded. Bias can be introduced if the outcomes assessed in the treatment arm and the external control arm differ based on the sources of data involved or the criteria used to establish outcomes. Therefore, assessing outcomes consistently across the treatment arm and the external control arm is essential for the results of an externally controlled trial to be credible.
Data Considerations For The External Control Arm
Data from another clinical trial used for an external control arm can provide more consistent data collection compared to using data collected during routine clinical care due to the use of defined protocols, provided that participant eligibility criteria, treatment administration, location of treatment sites, recording of associated medications, and assessments of adverse events and outcomes are similar. When the assessment and management of a disease changes over time, the lack of simultaneous data collection may be of particular concern and introduce bias.
The comparability of participant characteristics, timing and frequency of data collection, and patterns of care should be addressed when data collected on patients for non-research purposes as external control arms is utilized. Missing data sources obtained as part of routine clinical practice can threaten the validity of the results of an externally controlled trial.
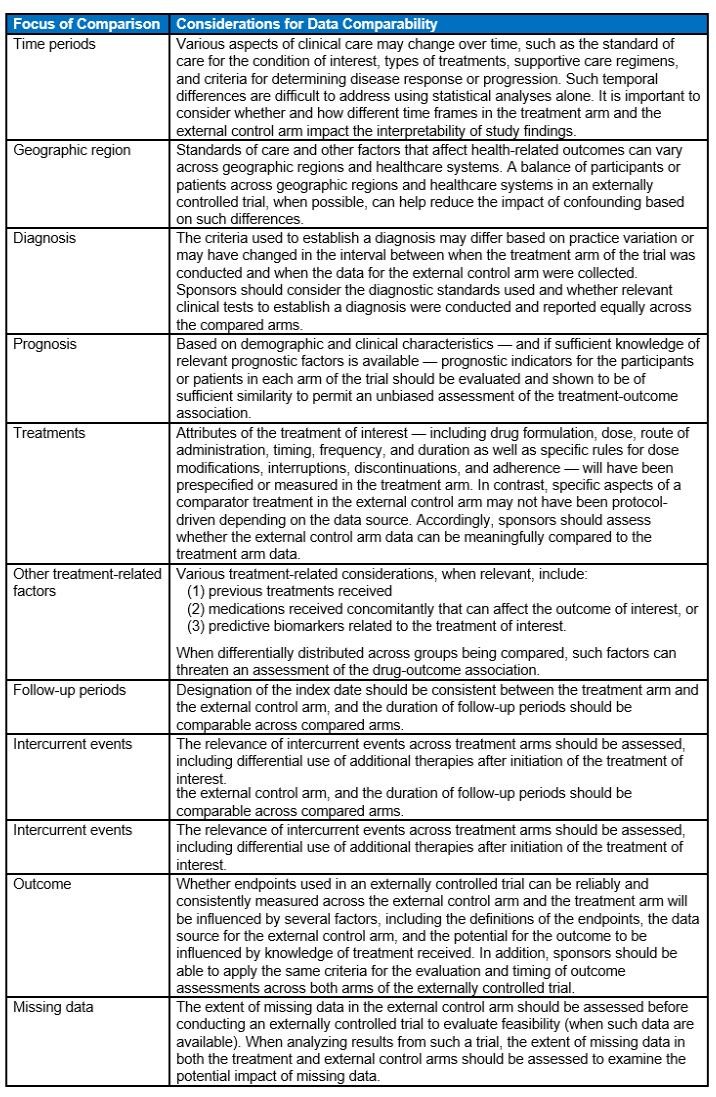
The following table provides a summary of key considerations regarding the comparability of data between the treatment arm and the external control arm. The considerations are directed at understanding and managing potential threats to the validity of externally controlled trials.
Analysis Considerations
A statistical analysis plan specifying the analyses of interest, calculations of statistical power, sample size, and plans to control the chance of erroneous conclusions should be documented before conducting an externally controlled trial. The statistical analysis plan with justification for the methods, strengths, and limitations of the statistical methods, and the study the protocol should be submitted to the FDA for review prior to enrollment of participants.
An externally controlled trial may not be an appropriate study design because of concerns of bias affecting the results when the anticipated effect size is modest. Sponsors should develop plans for assessing the impact of confounding factors and sources of bias with quantitative and/or qualitative analyses.
Externally controlled trials are susceptible to misclassification or mischaracterization of data. A biased assessment of the drug-outcome association may occur when data are misclassified or mischaracterized. The analytical methods should include a strategy for dealing with missing data, the potential impact of missing data, and potential bias caused by the missing data.
Considerations To Support Regulatory Review
Sponsors considering using an externally controlled trial instead of a randomized controlled trial should discuss this with the FDA early in a drug development program. Sponsors should provide a detailed description of the reasons why the proposed study design is appropriate, proposed data sources for the external control arm and an explanation of why they are fit for use, planned statistical analyses, and plans to address FDA’s expectations for the submission of data.
FDA regulations require sponsors to include in marketing applications relevant patient-level data, for both the treatment and external control arms and ensure the FDA has access to source documents and source data for the external control arm if requested.
Conclusion
Please submit written comments to Dockets Management Staff (HFA-305), Food and Drug Administration, 5630 Fishers Lane, Rm 1061, Rockville, MD 20852 or electronic comments to https://www.regulations.gov. Please reference docket number FDA-2022-D-2983 with all comments. Comments must be received by May 2, 2023. For questions regarding this draft guidance document, contact (CDER) Dianne Paraoan, 301-796-2500, or (CBER) Office of Communication, Outreach and Development, 800-835-4709 or 240-402-8010.
About The Author:
 Mark Allen Durivage has worked as a practitioner, educator, consultant, and author. He is managing principal consultant at Quality Systems Compliance LLC, an ASQ Fellow, and an SRE Fellow. Durivage primarily works with companies in the FDA regulated industries (medical devices, human tissue, animal tissue, and pharmaceuticals) focusing on quality management system implementation, integration, updates, and training. Additionally, he assists companies by providing internal and external audit support as well as FDA 483 and warning letter response and remediation services. He earned a BAS in computer aided machining from Siena Heights University and an MS in quality management from Eastern Michigan University. He holds several certifications, including CRE, CQE, CQA, CSSBB, RAC (Global), and CTBS. He has written several books available through ASQ Quality Press, published articles in Quality Progress, and is a frequent contributor to Life Science Connect. You can reach him at mark.durivage@qscompliance.com with any questions or comments.
Mark Allen Durivage has worked as a practitioner, educator, consultant, and author. He is managing principal consultant at Quality Systems Compliance LLC, an ASQ Fellow, and an SRE Fellow. Durivage primarily works with companies in the FDA regulated industries (medical devices, human tissue, animal tissue, and pharmaceuticals) focusing on quality management system implementation, integration, updates, and training. Additionally, he assists companies by providing internal and external audit support as well as FDA 483 and warning letter response and remediation services. He earned a BAS in computer aided machining from Siena Heights University and an MS in quality management from Eastern Michigan University. He holds several certifications, including CRE, CQE, CQA, CSSBB, RAC (Global), and CTBS. He has written several books available through ASQ Quality Press, published articles in Quality Progress, and is a frequent contributor to Life Science Connect. You can reach him at mark.durivage@qscompliance.com with any questions or comments.