
How Many Internal ClinOps Resources Should You Have For Study Startup?
 If you are like most small biotech companies, your budget is small and your timelines are as tight as possible in order to get the data needed to support the next round of funding and take the next step in your clinical development program. The best insurance you can use to achieve that next step and end up with a high-quality study that will pass an audit or inspection is proper planning ahead. This article will focus on proper resourcing of a sponsor’s internal clinical operations (ClinOps) team to adequately support a clinical trial during study startup.
If you are like most small biotech companies, your budget is small and your timelines are as tight as possible in order to get the data needed to support the next round of funding and take the next step in your clinical development program. The best insurance you can use to achieve that next step and end up with a high-quality study that will pass an audit or inspection is proper planning ahead. This article will focus on proper resourcing of a sponsor’s internal clinical operations (ClinOps) team to adequately support a clinical trial during study startup.
The Importance Of Right Resourcing
Proper resourcing is critical because:
- Our industry is highly regulated
- The stakes are high
- The competition is great
- The performance of the contract research organization (CRO) is not known in advance of contracting with them (even if you used the CRO before, you may have new team members, new processes, or new technologies to learn)
- Regulations and guidances change over time
- Change is a given
- Proper sponsor oversight of all trial activities is mandatory
The most important reason in the list above is ensuring proper sponsor oversight of all outsourced trial activities. In December 2016, the International Council for Harmonisation (ICH) adopted the revised E6 guideline, E6(R2), titled Integrated Addendum to Good Clinical Practice (GCP). The most extensive changes to ICH E6 were made to the sponsors section, beginning with a new section on quality management. The sponsor should “ensure oversight of any trial-related duties and functions carried out on its behalf, including trial-related duties and functions that are subcontracted to another party by the sponsor's contracted CRO(s).”1
A Lack Of Guidance
In an effort to understand how E6(R2) translates into actual day-to-day resourcing within ClinOps, I conducted an online search of “internal resourcing for clinical trials.” Most of what was available did not specifically address internal sponsor resourcing. What I did find was the following:
- A 2016 article by Lamberti, Chakravarthy, and Getz2 notes that, “despite new practices and process changes being instituted, the impact on improving study startup cycle times has been fairly small.”
- A 2017 article by McSpiritt3 notes that study startup “involves identifying the steps as well as who is going to perform the tasks within each process.”
- An online article by IQVIA4 states that “Despite efforts within the industry to finish trials on time, as many as 45 percent of clinical trials are completed late and approximately 80 percent of trials fail to meet their initial enrollment targets on time. As a result, there is a significant influence on cash flow and resource allocation, which can also impact other studies in a company’s development plan.
If resourcing is so vital, why isn’t it more widely discussed online and at conferences? I believe the answer lies in the view that sponsors spend a great amount of money identifying, qualifying, and then contracting with a CRO. They are paying the CRO to do the work, so why do they need to pay additional resources? The answer lies in oversight of the CRO.
When Does CRO Oversight Begin?
Oversight starts as soon as you begin to qualify and select your CRO and other vendors. Because every trial contains a number of pre-enrollment tasks — each of which is tightly linked to the previous and the next task — you can only run a successful trial if you are resourced appropriately to conduct those oversight tasks. There are simply some tasks that teams cannot “catch up on” once enrollment begins.
For example, for every study there are a minimum number of manuals and plans that must be authored, reviewed, and approved. These manuals and plans are the foundations of trial oversight. Even if the CRO provides the first draft, sponsors must review each one and approve it, often after several iterations of review. A sample list of such documents is below in Table 1.
Table 1: Sample List of Manuals and Plans Required For Study Startup

Critical Job Functions For CRO Oversight
Who in your organization will be qualified and resourced to review, approve, and oversee all the above items? You should have an experienced clinical project manager (CPM) who can oversee study startup and all of its required tasks, along with start and stop dates and task predecessors. This PM should create a complete timeline from drug product manufacturing through first subject enrolled that includes all the required tasks as well as related sub-tasks.
You also need one or more experienced clinical operations leads (COLs) to execute the various tasks within their areas of expertise, such as site selection, informed consent development, IRB/EC submissions, site budgets and contracts, site training/support, and CRA training and oversight.
C-Suite Blind Spot
I have observed that ClinOps resourcing in a small biotech company is something that is not appropriately or consistently planned for. I believe there are two main sources that influence this “blind spot:”
- C-suite executives that are running small biotech companies do not have the time or expertise to understand all the intricacies of study startup activities. Perhaps they came from large pharma where there were entire departments full of employees who conducted these activities. Now the executives are up close and personal with the intricacies and risks they have not been directly exposed to.
- Sponsor companies spend hundreds of thousands to millions of dollars outsourcing a clinical trial. It is very difficult to absorb and justify the cost of adding several internal resources to oversee the CRO.
Therefore, I wanted to develop a way to objectively quantify the internal sponsor ClinOps resources needed to successfully execute and oversee a trial.
Calculating The Number Of Full Time Equivalents You Need
There are approximately 115 tasks that have to be completed for any trial after the CRO has been selected and before the first subject is enrolled. The differences in resourcing for a Phase 1 study and a Phase 3 study are primarily driven by the number of sites and patients; however, the complexity of the protocol, the number of vendors supporting the trial, and the number of countries participating in the trial also play significant parts.
In order to quantify how many full time equivalents (FTEs) are required to adequately support the speed and quality needed for various types of trials, first outline all of the ClinOps tasks planned for startup for your particular study and then apply the number of human hours associated with each task. I would start with finite tasks, then assess ongoing tasks.
Finite Tasks
Finite tasks are those that have a start date and an end date. For example, a finite task might be authoring the original protocol version 1. Some sponsors will get a protocol synopsis to its final stage then hand it off to the CRO medical writer to author the first draft of the full protocol. Then there are rounds of review and editing until the protocol is final. In my experience, this can be one of the longest cycle times in startup because the protocol is the foundation for all aspects of the trial and what we “hang our hat on” in terms of the endpoints. In any case, hours for each author, reviewer, and other contributors need to be calculated.
To illustrate: If we tackle the list of manuals and plans listed above in Table 1, we can apply estimated required hours as shown in Table 2.
Table 2: Sample Estimated Hours Required for Manuals and Plans
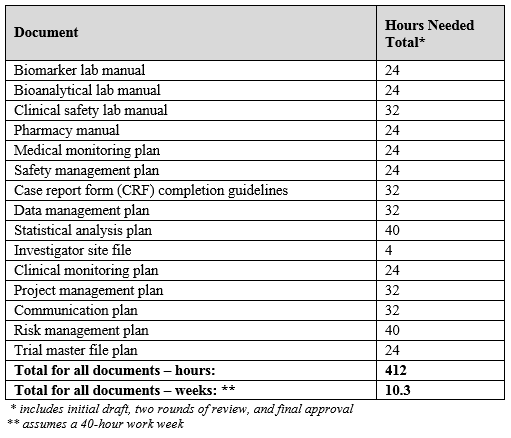
It appears that one or more ClinOps professionals can accomplish all of these tasks by dedicating 40 hours a week for 10 weeks collectively. However, not all tasks have one contributor, nor do they all start and end on the same date, so the 10 weeks can be distributed across the time from the final protocol through the first subject enrolled (and some of these manuals and plans can come after that milestone).
Ongoing Tasks
Ongoing tasks are those that repeat over time. This calculation includes listing each task, how long it takes, how often it occurs, and its duration. For example, the table below shows a sample of tasks and activities during a six-month startup period for a six-site study with a CRO on board:
Table 3: Sample Ongoing Tasks for a Six-Month Period in Startup*
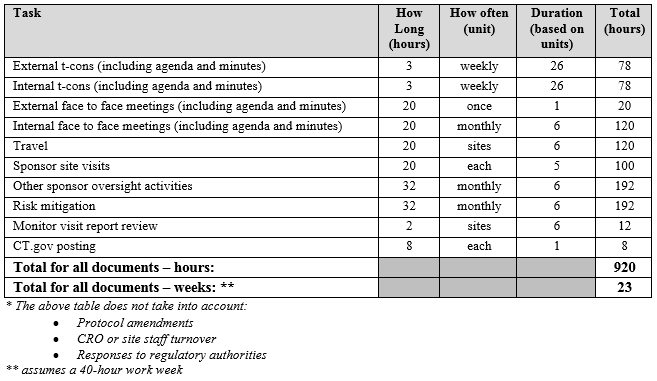
Here we see that one or more ClinOps professionals can accomplish all of these tasks by dedicating 40 hours a week for 23 weeks.
FTEs can be resourced by engaging two part-time consultants and then, as you approach larger, more complex trials, you can add to that pool as needed, perhaps by hiring full-time employees. You may want a quarter-time CPM and a half-time COL to start out with. Perhaps two half-time COLs can get the job done, which gives you back-up as well as added experience and skills.
Conclusion
Clinical trials are unpredictable, expensive, and complex. Sponsors need to resource appropriately, but it does not always happen correctly, and when it does not, it increases risk to the trial and to the sponsor.
There are many resourcing configurations and options that can be brought to bear, depending on the sponsor’s size, development plan, cash flow, and understanding of all of the above tasks and responsibilities in order to ensure proper sponsor oversight of all outsourced trial activities.
References:
- International Council for Harmonisation (ICH). 2016. Integrated Addendum to ICH E6(R1): Guideline for Good Clinical Practice E6(R2)
- Lamberti MJ, Chakravarthy R, Getz KA. “New Benchmarks for Trial Initiation Activities.” Applied Clinical Trials. Volume 25, Issue 12. December 1, 2016
- McSpiritt CM. “Why Are We Still Talking About Study Startup?” Clinical Leader. Oct 3, 2017. https://www.clinicalleader.com/doc/why-are-we-still-talking-about-study-startup-0001
- Archibald M, Brown M, Parmelee J, Turner N. “The Key to Successful Study Start-up: Right Path, Right Start, Right Patients.” https://www.iqvia.com/-/media/library/media-coverage/key-to-successfull-start-up-2020pharma.pdf
About The Author:
 Audrey Rossow is the owner of A Rossow Consulting, LLC, located in central Massachusetts. She has 25+ years’ experience in pharmaceutical and biotech clinical development, Phases 1 through 3b. Her core work is in project management and clinical operations. She is passionate about site engagement and support, patient recruitment and retention, and sponsor oversight of their CROs. She can be reached at audrey@arossowconsulting.com, and her website is http://www.arossowconsulting.com.
Audrey Rossow is the owner of A Rossow Consulting, LLC, located in central Massachusetts. She has 25+ years’ experience in pharmaceutical and biotech clinical development, Phases 1 through 3b. Her core work is in project management and clinical operations. She is passionate about site engagement and support, patient recruitment and retention, and sponsor oversight of their CROs. She can be reached at audrey@arossowconsulting.com, and her website is http://www.arossowconsulting.com.