
How To Combine Quality Management With Risk-Based Monitoring In Clinical Trials
By Meaghan Powers, lead consultant, Halloran Consulting Group

The research community was introduced to risk-based monitoring (RBM) in 2013, when the FDA published its industry guidance, “Oversight of Clinical Investigations – A Risk-Based Approach to Monitoring.” This guidance tasked industry sponsors with adopting a formal approach to quality management by embracing technology and leveraging access to real-time information to drive a more structured approach to risk in study conduct, of which monitoring is a critical element. To be successful, clinical trial sponsors and contract research organizations (CROs) need to comply with the guidance, proactively manage risks, and keep their trials on track and on budget. That being said, it is important to understand why RBM alone is not enough for a trial to succeed and what the industry can do to extend the use of risk-based strategies to ultimately support improved data quality and increased patient safety in clinical trials.
So, what is driving the change in how a clinical trial is conducted? Despite the regulatory guidance documents available on this topic, the current uptake of risk-based approaches to monitoring clinical trials is still lagging as industry sponsors are challenged with working toward transparency and integrating quality into the clinical trial life cycle. To facilitate this adjustment, risk-based quality management (RBQM), a process that focuses on quality by design throughout the clinical trial by viewing it through a life cycle lens, was introduced. No longer is quality accomplished by way of checklists and box checking, but instead by integrating steps that are specific and measurable. ICH E8 R1 reminds us “Quality should rely on good design and its execution rather than overreliance on retrospective document checking, monitoring, auditing, and inspection.” Quality should be discussed openly, and issues such as poor trial design, misconduct, and data collection should be deliberated to avoid future occurrences. Companies can only work well with a vendor or internal quality partners if they have a measurable deliverable, not a stack of documents (SOPs/plans) that describe a perfect state that is not achievable. Yet, even with this ambitious approach, there are several gaps in how RBQM principles are enforced, including: risk characterization processes aren’t applied consistently; issue management among many organizations does not allow for frequent feedback; risk communication and reporting are still mostly manual; tracking/querying is inconsistent; risk review is constrained by incomplete analytics; and risk control key risk indicators (KRIs) are lacking robustness and trial specific capabilities.
3 Key Steps To Successful Execution
RBM is an important component of how an overall RBQM strategy is executed, so it is important that we understand what enhancements can be applied to strengthen the likelihood of success. A big part of this is how RBM is executed with increased use of electronic systems and improvements in statistical assessments, which present the opportunity for evolved monitoring approaches. RBM has become an established form of monitoring from regulatory agencies and will soon become the norm. With that, the FDA has laid out three necessary steps to be taken in sequence to ensure RBM and RBQM success:
- First, a risk assessment must be conducted both pre-study and ongoing during the trial.
- Second, a well-articulated study protocol should be developed based on factors identified during this risk assessment.
- Finally, an RBM approach should be tailored based on both the risk assessment and the protocol.
This sequence allows for focus on risks to the most critical data elements and the highest risk areas, with earlier detection of potential safety issues and trends by incorporating centralized monitoring and analytics. The focus has changed from source data verification (SDV) and transcription to a more process-based mindset. When looking at monitoring through an RBQM lens, it correlates to more critical thinking (moving from a risk-based approach to quality management), technology supporting real-time data access and new data review and analytic approaches, and sponsors expecting earlier insights from their CROs.
Historically, once a risk or issue was identified in a study, the sponsor and CRO went back to training or planning as a result of failures, which results in more SOPs and plans. The challenge is to study the cause of the failure and change the approach using models such as the Plan-Do-Study-Act Model (outlined in Figure 1), which can be key to establishing a foundation for critical thinking from the initial protocol design and development step. Models such as these help establish pathways for triggering risk signals, identify if/then scenarios for key risks, and recruit key players engaged with KRIs to empower their ability to identify risk.
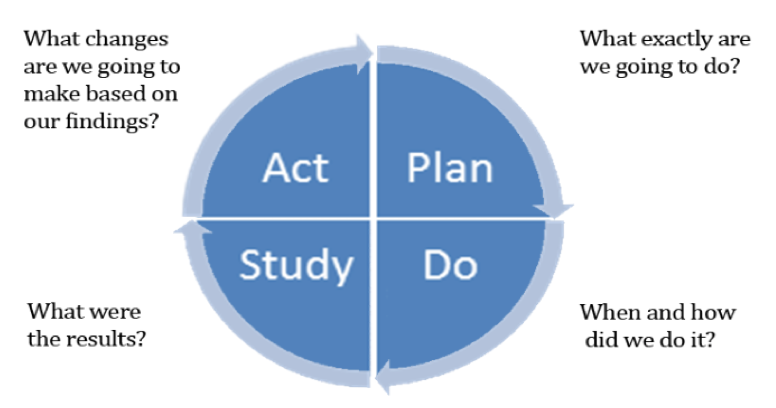
Figure 1: Plan-Do-Study-Act Model
Filling in the gap between where we have been and the way forward requires addressing the time between protocol risk assessment and monitoring conduct — also referred to as the integrated strategic monitoring plan (ISMP). ISMP defines the relationship between critical variables and the monitoring approach. It also enables trial specificity in the monitoring plan to ensure safety, data integrity, and compliance. Starting with a risk assessment and moving through to a well-designed investigational plan based on that risk assessment is the main starting point for success. Combining a risk-based investigational plan with ISMP will lead to the successful deployment of the RBM strategy. It is important to remember that risks change over time — the process of RBM is not static, so neither is the ISMP.
Impact On The Trial Site
But let us not forget about the clinical site in all this. At the site level, the integration of RBQM likely feels similar to the change the industry experienced with the shift from paper to electronic data capture (EDC). The adoption of EDC was slow at first, but as the benefits became clear, it became the norm across most clinical sites. Central monitoring, which is at the core of RBQM model at the site level, will likely have a similar path. Central monitoring is the continuous review of data and risks to drive study performance and is often used in the RBM model. Central monitoring uses holistic assessments of clinical trial data, utilizing analytics and visualizations to detect trends, outlier data, and areas of emerging risks, which aligns with the overall RBQM approach. This can lead to early issue detection and resolution, as well as a targeted review of sites that need the most attention, resulting in enhanced patient safety and improved data integrity. Central monitoring should reduce unnecessary monitoring while increasing quality. Sponsors should prepare site investigators in advance for vastly different monitoring frequencies and methodologies and be prepared to explain the rationale for such changes. Sites must be on board with the RBM approach in order to be successful. Transparency, through the direct and indirect effects of RBM, will not only increase efficiency and data quality, but also help shape a safer future for the clinical trial participant.
Conclusion
Industry needs to keep the engine moving forward on RBQM. Change and continuous improvement are key contributors that can lead to enhanced quality, effectiveness, and efficiency. It is critical to be able to measure success in your RBQM strategy to demonstrate if, as a company, you are where you want to be. If you’re not looking at data and metrics, it will be tough to answer that question. Each company’s journey will be driven by many factors (culture, interpretation, disease state focus, change load, etc.), so stay focused on the why; keep it simple, communicate, and don’t over engineer.
About The Author:
 Meaghan Powers, lead consultant for clinical operations at Halloran Consulting Group, brings more than 17 years of experience in clinical trial execution and project management, across both the pharmaceutical and medical device spaces. She has significant experience in both cardiology and urology/pelvic health, utilizing her experience of clinical trial design, execution, and process improvement to drive transformation. Prior to joining Halloran, Powers was at Boston Scientific, where she successfully managed programs through the project life cycle of clinical study design and study publication. Before Boston Scientific, she worked as a clinical trial coordinator at Dana Farber Cancer Institute.
Meaghan Powers, lead consultant for clinical operations at Halloran Consulting Group, brings more than 17 years of experience in clinical trial execution and project management, across both the pharmaceutical and medical device spaces. She has significant experience in both cardiology and urology/pelvic health, utilizing her experience of clinical trial design, execution, and process improvement to drive transformation. Prior to joining Halloran, Powers was at Boston Scientific, where she successfully managed programs through the project life cycle of clinical study design and study publication. Before Boston Scientific, she worked as a clinical trial coordinator at Dana Farber Cancer Institute.