
How to Optimize Success in Clinical Trials of Antidepressants
By Paula Jacobsen, Ph.D., and Nick DeMartinis, M.D., Praxis Precision Medicines

This is an exceptionally exciting time to be developing therapies for patients suffering from depressive disorders. Advances in antidepressant pharmacology have led to diverse and promising therapeutic options. However, it is important to acknowledge the challenges of conducting clinical trials for major depressive disorder (MDD).
Chief among these challenges is the sensitivity of clinical trial methodology for demonstration of efficacy in MDD. A meta-analysis of antidepressant programs that included FDA-approved antidepressant clinical trial data from randomized, double-blind, placebo-controlled acute treatment studies conducted between 1995 and 2008 revealed that only 50% were successful despite the inclusion of only studies that evaluated doses in the approved therapeutic range.1 The clinical trials that failed did not demonstrate efficacy even though the study drug was effective in treating the condition under investigation. This high percentage of failed clinical trials reflects the challenge of developing effective new drugs for MDD, primarily due to the subjective nature of assessments and robust placebo response. In order to bring promising therapies safely, effectively, and expediently to the millions of individuals who need them, we need to reevaluate learnings from the past few decades to overcome the challenges in design and conduct of clinical trials for antidepressant drugs.
Study Design And Protocol
Diagnostic Considerations
Appropriate participant selection is core to the success of a clinical trial for MDD. Sponsors can accomplish this by standardizing the diagnostic process using structured psychiatric interviews, which can be used to thoroughly assess eligibility requirements and limit variability in patient characteristics. The probability of success in clinical trials of antidepressants can be increased by requiring a diagnosis of recurrent depression.2 This is because those who experience their first depressive episode tend to be more likely to respond to placebo, which makes signal detection challenging.3 Participants who have experienced at least three depressive episodes, but are not treatment resistant, are ideal candidates for MDD trials because they tend to have the greatest baseline severity and are least likely to spontaneously improve.4
In order to ensure quality control during the enrollment process, it is important to have a system in place to confirm strict adherence to inclusion and exclusion criteria, which frequently involves external surveillance of key primary efficacy assessment ratings via review and co-rating of recorded assessment interviews.5 This approach also supports ongoing rater quality assurance and feedback to optimize consistency in administration and scoring of the subjective assessments used to determine efficacy. An additional degree of homogeneity and likelihood of drug-placebo separation can be gained through a process by which participants are additionally assessed by an independent and blinded expert interview focusing on both eligibility confirmation and evaluation of characteristics that decrease the potential for placebo response. In a review of nine trials of MDD and treatment resistant depression, Freeman et al found that approximately one out of seven participants who were included in the studies by on-site investigators did not meet enrollment criteria following an externally administered questionnaire. Furthermore, in sites where enrollment was monitored by this external adjudication, the placebo response rates dropped to 13% to 27%, significantly below the average of 35% to 40% seen in psychiatric clinical trials.6
Baseline Severity
In their review of antidepressant trials, Khin and colleagues found that baseline severity was a better predictor of a successful outcome than trial duration, location, or sample size.1 In general, there is greater room for signal detection in patients with moderate-to-severe MDD symptoms compared to those with mild symptoms because participants with lower baseline severity tend to be high placebo responders.7 Including participants with moderate MDD is prudent, because if the inclusion criteria are restricted to severe disease, there may be a greater tendency toward baseline inflation of depression severity by raters in order to recruit enough participants.8 While this factor can be addressed through the process of independent review of rating interview recordings,9 it is also important to include moderate severity MDD participants to support generalizability to the clinical population of MDD patients warranting pharmacological treatment.
The Issue of Intentionally Non-Adherent Patients
By participating in clinical trials, individuals can gain access to current potential treatments and receive compensation for their participation. The majority of patients who take part in MDD trials are highly valued participants who have made personal sacrifices in order to contribute to the development and availability of new antidepressants. A small but significant minority of intentionally non-adherent patients may use misleading tactics to enroll in multiple trials primarily for financial gain. By misrepresenting symptoms or failing to report concurrent medication or co-morbid disorders, intentionally non-adherent patients can corrupt the quality of data collected, which can diminish effect size, contribute to trial failure, and potentially alter the safety profile of the drug under study.10
To mitigate this issue, proactively screening participants through clinical trial registries that use common identifiers can identify those who attempt to enroll in more than one trial, as well as those who have participated in previous trials with an exclusionary diagnosis.10
Efficiency of Protocol Designs
Increasing the complexity of a trial design in an attempt to enhance signal detection can have counterintuitive impacts that result in the opposite effect. Protocols with a large number of assessments have been found to be more likely to result in an increased placebo response.8, 11 Importantly, a simple protocol focused on maintaining the integrity of the primary efficacy analysis can be more effective at evaluating a treatment effect while also decreasing the burden of participation for participants and decreasing the operational burden on the research team.12 Trial methodology should generally not be overly complicated in an attempt to mitigate the placebo effect. A number of design strategies intended to reduce the placebo response have been employed with mixed results. Extending the length of the trial to accommodate the fluctuating nature of depression, for example, has not been successful.1, 13 There have been some study designs specifically created to reduce placebo response, such as the Sequential Parallel Comparison Design (SPCD)14 and the Run-in with Adherence Monitoring for Prequalification but Undiminished Participation (RAMPUP)15 design, and all have had some success but with varying degrees of methodological complexity and some mixed results as well.
Choice Of Assessments
Well-designed antidepressant trials should use a single primary depression scale as the primary endpoint, and the number of assessments and patient-reported outcomes (PROs) should be limited to only what is needed to answer the key scientific objectives. Currently, the standard assessments used in clinical trials for antidepressants in adults are the Hamilton Rating Scale for Depression (HAM-D) and the Montgomery-Asberg Depression Rating Scale (MADRS).16 Other scales, such as HAM-D6, can be used as supplements, but the HAM-D and MADRS are best for trials of novel antidepressants because they assess a wide range of symptoms. These tools are equally effective, so the best practice is to choose the one best suited to evaluate the therapy under investigation and to avoid using additional assessments when possible. Inclusion of multiple assessments can skew results to either increase the chance of false positives or increase the placebo response and blunt signal detection.17
Sample Size And Power Considerations
The most successful clinical trials are both adequately powered12 and have a sufficient proportion of placebo-treated participants for the confident detection of drug-placebo differences.18 For trials in which the effect size can be expected to be small, a large number of participants are required to demonstrate the hypothesis that there is indeed a difference between those who receive active treatment and those who do not. However, one must take care to ensure that selection of appropriate participants is not compromised when trying to reach recruitment targets. In an analysis of Phase 3 clinical trials of paroxetine, for example, Liu et al showed an increase in placebo responders among those who were enrolled later in the trial.19 There are a number of examples in which the combination of study size and recruitment timeline pressure results in failure to detect efficacy for a drug that is truly effective.20
Additionally, the delicate balance between overpowering and underpowering the study must be matched with an adequately powered placebo arm. When the size of the placebo cohort is substantial, the placebo response is more predictable, which allows for better signal detection in the study overall.2, 12 The magnitude of the placebo response also provides useful information. In an analysis of 17 successful clinical trials of recently approved antidepressant drugs, the average placebo response at two weeks was an approximately six-point decrease in the HAM-D17 total score (Figure 1).21 This data is useful in understanding the magnitude of an acceptable level of placebo response associated with successful MDD trials.
Figure 1. Mean Change from Baseline at Day 14 for Placebo Group by Study
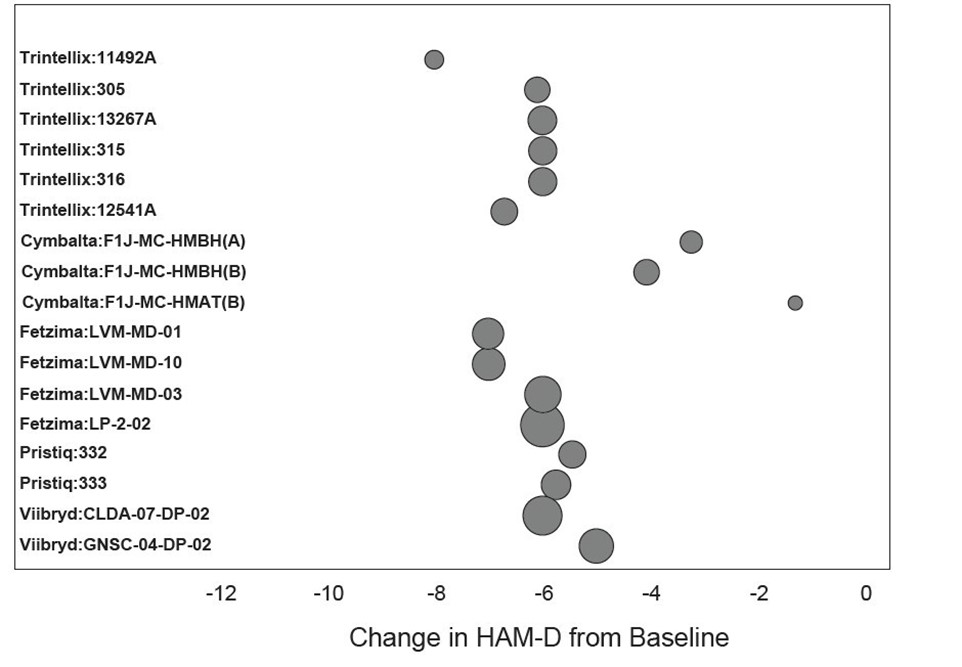
Change in baseline to day 14 in HAM-D total score for the placebo arm of selected randomized placebo-controlled studies of recently approved antidepressants. Data was taken from acute studies which demonstrated statistically significant improvement over placebo. Bubble size is proportional to the sample size of the placebo group in each study. Across studies, the sample size in the placebo group ranged from 89 to 277.
Source: Brintellix/Trintellix Summary Basis of Approval (SBA) 2013, Viibryd SBA 2011, Cymbalta SBA 2011, Fetzima SBA 2013, Oleptro SBA 2010, Pristiq SBA 2008
Study Conduct
Participant Education On Placebo-Controlled Trials
Participant education is an integral part of the clinical study process, as it controls patients’ expectations, increases adherence to study protocols, and helps to mitigate the placebo response. It is important that patients understand that they do not need to get better to remain in the study.22 Education can be achieved by using carefully crafted scripts such as the Placebo-Control Reminder Script, providing educational videos, and ensuring that participants are informed about what it means to be in a clinical trial.22 While many experienced research sites attend to patient education, standardization of an approach across all sites can maximize the benefit and help minimize variability in efficacy endpoints.
Sponsor Engagement
Lastly, it is important for sponsors, sites, and the clinical research organization (CRO) to maintain an active and collaborative relationship to ensure trial success. When sponsors and CROs are actively engaged in the participant enrollment process and clinical investigation, sites can be motivated to prioritize quality candidate enrollment and research over meeting quotas and deadlines.6, 11 In order to optimize this benefit, however, it is critically important that sites receive consistent messages supporting that imperative from both the CRO and sponsor.
Table 1: Key principles of a successful study.
| Principle | How | Why |
|
Strict adherence to eligibility criteria for participant selection. |
Use structured interviews. Consider external adjudication. Use subject registry database to detect intentionally non-adherent patients. |
Inclusion of appropriate participants who are accurately diagnosed will produce high-quality results with reliable external validity. Placebo response rates drop with structured interviews and external oversight.6 |
|
Uncomplicated protocol design. |
Implement a straight-forward double-blind placebo-controlled protocol without additional complexities such as placebo lead-ins or multiple arms for Phase 3 studies. |
Mitigates placebo effect. Decreases site burden. Produces unambiguous data. |
|
Limit the number of assessments and/or time points for assessments to those necessary to answer the key questions. |
Limit redundant scales, e.g., choose either HAM-D or MADRS but not both throughout the study. Limit the number of secondary and exploratory assessments. |
Maintains focus on primary and key secondary endpoints. Mitigates the placebo effect. |
|
Include a robust placebo cohort. |
Adequately power the placebo arm of the trial, but do not overpower. Effect size must still be clinically meaningful. |
Increases the predictability of the placebo response. Mitigates participant expectation. Improves signal detection.23 |
|
Educate participants about being in a placebo control trial. |
Use a placebo control script or other patient-facing placebo mitigation strategy. |
Manages participant expectations. Decreases the placebo response.22 Improves adherence, which increases data quality. |
|
Monitor medication adherence and have a system to intervene if needed. |
Use adherence monitoring technology. |
Increases the reliability and validity of the results by increasing adherence to the medication protocol, and identifying non-adherence early. |
|
Use experienced sites and research-focused environments. |
Keep metrics on sites. Monitor sites. Have a system for remediation. |
Mitigates the placebo effect. Ensures consistency of data. |
|
Ensure rater consistency and calibration. |
Utilize a rater training and data monitoring vendor. Use site-based raters with external oversight. |
Reduces baseline inflation. Reduces expectation bias. Increases reliability of data.26 |
|
Maintain a spirit of scientific collaboration. |
Foster ongoing relationships between sponsor, CRO, and sites. |
Shared investment in scientific inquiry speeds progress. |
|
Commit to publishing negative results. |
In journals On clinicaltrials.gov On company website |
Others in the scientific community can learn from failures as well as successes. |
Conclusion
By taking the time to reevaluate the methodology and results of prior studies, distilling best practices, and maintaining discipline in prioritizing factors that support the achievement of a study’s scientific objectives, we can substantially increase the chances of success beyond the 50% rate observed in the broad range of prior MDD clinical trials. With almost 300 million individuals today who suffer from depression,27 it is essential that we use our learnings to expand the treatment landscape to evaluate and deliver new and effective medicines to improve the lives of people with MDD and those around them. If you would like to learn more, you can read our full white paper here.
References
- Khin NA, Chen YF, Yang Y, Yang P, Laughren TP. Exploratory analyses of efficacy data from major depressive disorder trials submitted to the US Food and Drug Administration in support of new drug applications. J Clin Psychiatry. Apr 2011;72(4):464-72. doi:10.4088/JCP.10m06191
- Rutherford BR, Roose SP. A model of placebo response in antidepressant clinical trials. Am J Psychiatry. Jul 2013;170(7):723-33. doi:10.1176/appi.ajp.2012.12040474
- Sonawalla SB, Rosenbaum JF. Placebo response in depression. Dialogues Clin Neurosci. Mar 2002;4(1):105-13.
- Rapaport MH, Maddux RE. Challenges in the development of clinical trials for major depressive disorder: lessons learned from trials in minor depression. Dialogues Clin Neurosci. Dec 2002;4(4):402-7.
- Targum SD, Pendergrass JC, Toner C, Asgharnejad M, Burch DJ. Audio-digital recordings used for independent confirmation of site-based MADRS interview scores. Eur Neuropsychopharmacol. Nov 2014;24(11):1760-6. doi:10.1016/j.euroneuro.2014.08.016
- Freeman MP, Pooley J, Flynn MJ, et al. Guarding the Gate: Remote Structured Assessments to Enhance Enrollment Precision in Depression Trials. J Clin Psychopharmacol. Apr 2017;37(2):176-181. doi:10.1097/JCP.0000000000000669
- Holmes RD, Tiwari AK, Kennedy JL. Mechanisms of the placebo effect in pain and psychiatric disorders. Pharmacogenomics J. Nov 2016;16(6):491-500. doi:10.1038/tpj.2016.15
- Potter WZ, Mallinckrodt, C.H., Detke, M. Controlling placebo response in drug development: lessons learned from psychopharmacology. Pharmaceutical Medicine. 2014;28:53-65. doi:10.1007/s40290-014-0052-8
- Targum SD, Catania CJ. Audio-digital recordings for surveillance in clinical trials of major depressive disorder. Contemp Clin Trials Commun. Jun 2019;14:100317. doi:10.1016/j.conctc.2019.100317
- Shiovitz TM, Bain EE, McCann DJ, et al. Mitigating the Effects of Nonadherence in Clinical Trials. J Clin Pharmacol. Sep 2016;56(9):1151-64. doi:10.1002/jcph.689
- Marder SR, Laughren T, Romano SJ. Why Are Innovative Drugs Failing in Phase III? Am J Psychiatry. Sep 1 2017;174(9):829-831. doi:10.1176/appi.ajp.2017.17040426
- March JS, Silva SG, Compton S, Shapiro M, Califf R, Krishnan R. The case for practical clinical trials in psychiatry. Am J Psychiatry. May 2005;162(5):836-46. doi:10.1176/appi.ajp.162.5.836
- Furukawa TA, Cipriani A, Atkinson LZ, et al. Placebo response rates in antidepressant trials: a systematic review of published and unpublished double-blind randomised controlled studies. Lancet Psychiatry. Nov 2016;3(11):1059-1066. doi:10.1016/S2215-0366(16)30307-8
- Fava M, Evins AE, Dorer DJ, Schoenfeld DA. The problem of the placebo response in clinical trials for psychiatric disorders: culprits, possible remedies, and a novel study design approach. Psychother Psychosom. May-Jun 2003;72(3):115-27. doi:10.1159/000069738
- McCann DJ, Petry NM, Bresell A, Isacsson E, Wilson E, Alexander RC. Medication Nonadherence, "Professional Subjects," and Apparent Placebo Responders: Overlapping Challenges for Medications Development. J Clin Psychopharmacol. Oct 2015;35(5):566-73. doi:10.1097/jcp.0000000000000372
- FDA. Guidance for Industry: Major depressive disorder: developing drugs for treatment. Revision 1 ed. Silver Springs, MD: US Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER); June, 2018.
- Leon AC. Implications of clinical trial design on sample size requirements. Schizophr Bull. Jul 2008;34(4):664-9. doi:10.1093/schbul/sbn035
- Kobak KA, Leuchter A, DeBrota D, et al. Site versus centralized raters in a clinical depression trial: impact on patient selection and placebo response. J Clin Psychopharmacol. Apr 2010;30(2):193-7. doi:10.1097/JCP.0b013e3181d20912
- Liu KS, Snavely DB, Ball WA, Lines CR, Reines SA, Potter WZ. Is bigger better for depression trials? J Psychiatr Res. Jul 2008;42(8):622-30. doi:10.1016/j.jpsychires.2007.07.003
- Publishing negative clinical trials results: the case of reboxetine. Nurse Prescribing. 2010;8(11):516-518. doi:10.12968/npre.2010.8.11.79783
- Form S-1 Registration Statement for Praxis Precision Medicines, Inc. (2020).
- Cohen EA, Hassman HH, Ereshefsky L, et al. Placebo response mitigation with a participant-focused psychoeducational procedure: a randomized, single-blind, all placebo study in major depressive and psychotic disorders. Neuropsychopharmacology. Mar 2021;46(4):844-850. doi:10.1038/s41386-020-00911-5
- Papakostas GI, Fava M. Does the probability of receiving placebo influence clinical trial outcome? A meta-regression of double-blind, randomized clinical trials in MDD. Eur Neuropsychopharmacol. Jan 2009;19(1):34-40. doi:10.1016/j.euroneuro.2008.08.009
- Bain EE, Shafner L, Walling DP, et al. Use of a Novel Artificial Intelligence Platform on Mobile Devices to Assess Dosing Compliance in a Phase 2 Clinical Trial in Subjects With Schizophrenia. JMIR Mhealth Uhealth. Feb 21 2017;5(2):e18. doi:10.2196/mhealth.7030
- Getz K, Smith Z, Shafner L, Hanina A. Assessing the Scope and Predictors of Intentional Dose Non-adherence in Clinical Trials. Ther Innov Regul Sci. Nov 2020;54(6):1330-1338. doi:10.1007/s43441-020-00155-x
- Kobak KA, Brown B, Sharp I, et al. Sources of unreliability in depression ratings. J Clin Psychopharmacol. Feb 2009;29(1):82-5. doi:10.1097/JCP.0b013e318192e4d7
- Depression. Website. World Health Organization. Accessed January 29, 2021. https://www.who.int/en/news-room/fact-sheets/detail/depression
About the Authors:
 Paula Jacobsen is the senior director of clinical development at Praxis Precision Medicines and currently serves as the clinical science lead for Praxis’ depression program. She has more than 20 years of experience in the pharmaceutical industry, providing leadership and expertise in trial design, protocol development, and study execution. Additionally, she is one of the founding members of the CNS Summit. She holds a Ph.D. in research methodology from Loyola University Chicago, an M.S. in biology from the University of Texas, and a B.S. degree from North Central College.
Paula Jacobsen is the senior director of clinical development at Praxis Precision Medicines and currently serves as the clinical science lead for Praxis’ depression program. She has more than 20 years of experience in the pharmaceutical industry, providing leadership and expertise in trial design, protocol development, and study execution. Additionally, she is one of the founding members of the CNS Summit. She holds a Ph.D. in research methodology from Loyola University Chicago, an M.S. in biology from the University of Texas, and a B.S. degree from North Central College.
 Nick DeMartinis, M.D., is the vice president of clinical development at Praxis Precision Medicines. He has over 20 years of clinical research and drug development experience in academia and industry, including 13 years as an academic investigator in numerous clinical trials across 10 psychiatric indications at Penn and the UConn School of Medicine. He holds an M.D. from the Medical College of Georgia and completed his residency in psychiatry and fellowship in mood and anxiety disorders at the University of Pennsylvania School of Medicine. He holds a B.Ch.E. degree from Georgia Institute of Technology.
Nick DeMartinis, M.D., is the vice president of clinical development at Praxis Precision Medicines. He has over 20 years of clinical research and drug development experience in academia and industry, including 13 years as an academic investigator in numerous clinical trials across 10 psychiatric indications at Penn and the UConn School of Medicine. He holds an M.D. from the Medical College of Georgia and completed his residency in psychiatry and fellowship in mood and anxiety disorders at the University of Pennsylvania School of Medicine. He holds a B.Ch.E. degree from Georgia Institute of Technology.