
Incorporating Patient-Centric Outcome Metrics In Rare Disease Trials
By Claudia Dall’Osso, Ph.D., and Ian Love, Ph.D., Decision Resources Group (DRG)

With a hyper focus on validated endpoints, developers often fail to capture outcome measures important to patients
Commercial viability of pharmaceuticals depends on getting medicines to patients, which requires developers to evaluate investigative therapies using outcomes that are quantifiable, easy to interpret, and clinically relevant. However, clinical relevance is often a matter of stakeholder perspective: what developers view as meaningful may not appear as such to patients and caregivers.
Historically, developers have chosen endpoints often without carefully considering their relevance to patients or caregivers; however, in the last few years, patients’ perspectives in the drug development process have become more prominent, and drug companies and regulatory agencies are increasingly working to incorporate patient-centric outcomes in clinical trials. Not to be confused with patient-reported outcomes (PROs), which are simply outcomes directly reported by patients, patient-centric outcomes are clinical metrics that matter to patients and/or caregivers.
In rare disease markets, the decision to incorporate patient-centric outcomes can prove to be the difference between the success and failure of a development program. At the recent National Organization for Rare Disorders (NORD) summit1, FDA officials reported that, among rare disease trials that ultimately failed, one of the most common complaints of participants was that the trial neglected to capture the positive outcomes they were experiencing. A recent example of this phenomenon involves Sanfilippo syndrome, a rare lysosomal storage disease characterized by cognitive regression in which loss of sleep and organ inflammation are also common. Although sleep loss has a tremendous negative impact on patients’ quality of life, developers typically measure the impact of emerging therapies on cognitive decline. Indeed, per DRG research conducted with patient advocacy executives, patient feedback collected by the CureSanFilippo foundation suggested that failed clinical programs, while unable to slow cognitive regression, were improving sleep loss. From the caregivers’ perspective, preventing sleep loss alone was a meaningful enough outcome to support adoption of the agent by SanFilippo patients. Additionally, patient advocacy groups, a powerful voice in rare disease markets, and treatment providers, who are tasked with the day-to-day management of patients, are more likely to push for the adoption of medications that they believe address some patient needs, even if they miss the primary endpoint set forth in the trial.
How Should Developers Approach Patient-Centric Outcome Metrics?
In most rare disease indications, there are no established patient-centric endpoints. To include such endpoints in clinical trials, manufacturers must first identify specific disease manifestations that negatively impact patients’ lives and assess the magnitude of change that constitutes a meaningful improvement to patients. After determining what is important to measure, manufacturers need to develop and validate a method to collect the outcome measure and to collaborate with key stakeholders, including patients and patient advocacy groups, regulatory agencies, payers, and physicians, to establish a path to regulatory approval, uptake, and reimbursement.
Patient Engagement
The earliest stages of the process to develop patient-centric outcomes require an in-depth analysis of the patient journey, key pain points, and unmet needs. Developers must gain a comprehensive understanding of the difficulties that patients and caregivers face and identify outcomes that would represent meaningful improvements in the daily lives of these people. A complicating factor is that, in some cases, the natural history of the disease is not fully known: developers must also assess disease progression to be able to design outcome metrics meaningful to patients in different stages of the disease.
The most straightforward method for manufacturers to gauge patients’ perspectives is by engaging patient advocacy groups or other entities that allow for direct interaction with patients (e.g., social media platforms). Patient advocacy groups typically have strong ties with their patient communities and have often invested resources to better understand the natural history of the disease. DRG research indicates that these organizations are willing to collaborate with developers by offering their expertise on disease progression as well as support for conducting patient listening activities. Online discussion forums/platforms can also provide valuable insight into patients’ perspectives: for example, DRG’s social intelligence research, which captures the patient perspective and mind-set by gleaning and collecting patient conversations from online social platforms, has proved to be a critical market research channel for marketers and developers. Moreover, several disease-specific platforms designed to facilitate peer to peer connections among patients are emerging, and these tools could also support patient engagement from a manufacturer standpoint. Regardless of how it is collected, patients’ engagement can provide much-needed context to patient journeys compiled through analysis of real-world evidence and/or physicians’ opinions.
It is worth noting that many patient advocacy groups maintain close relationships with expert clinicians and communicate regularly with regulatory agencies. Such connections can be particularly helpful for developers looking to gain wide acceptance of novel clinical trial endpoints. The FDA’s ongoing effort to elicit patient feedback through interaction with patient organizations is a testament to the rising importance of these groups in the drug development process. In 2017, in collaboration with the Clinical Trial Transformation Initiative and patient advocacy organizations, the agency created the Patient Engagement Collaborative, a forum designed to explore how to achieve more meaningful patient engagement in medical product development. Furthermore, within the rare disease space, the FDA has adopted a memorandum of understanding with NORD and has planned a series of patient listening sessions, with the goal of discussing unmet needs and informing drug developers. At the recent NORD summit,1 the agency announced a plan to create a series of guidance documents on “the science of patient engagement” and how best to conduct this type of research.
Method Validation
Once developers have defined the outcomes that are important to patients and understand the effect size necessary to demonstrate a meaningful difference to patients, they must develop and validate methods to assess these outcomes. This process can be time-consuming and expensive, and it may not always be straightforward.
An example of a novel patient-centric outcome metric is the multi-luminance mobility test (MLMT), developed by Spark Therapeutics as a method to assess Luxturna’s efficacy for inherited retinal disease (IRD) caused by biallelic RPE65 mutations. The need for the test stemmed from the developer’s realization that the traditional visual acuity tests measure eye function, but not visual function (i.e., how patients function while performing vision-dependent activities). Luxturna’s target patients, especially in the early stages of the disease, struggle to see in low-light conditions and consequently have difficulties navigating poorly lit environments. However, traditional visual acuity tests are carried out under normal light conditions (usually a well-lit physician’s office) and fail to capture an important aspect of the disease. Thus, Spark developed a novel multi-luminance mobility test (MLMT) to quantitatively gauge patients’ ability to navigate their environment at varying light levels — a meaningful way to simulate the daily challenges of IRD patients.2
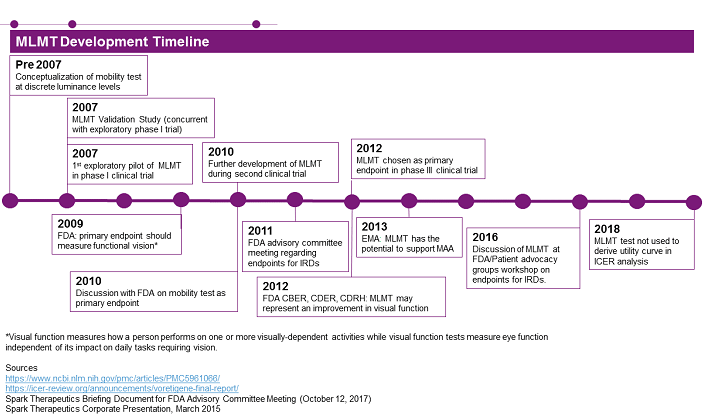
Figure 1
Figure 1 summarizes the development timeline for the MLMT test, showing how it was conceptualized, validated, and implemented as the primary endpoint in the pivotal Phase 3 clinical trial for Luxturna. Furthermore, company-released material highlights how Spark worked collaboratively with both the FDA and EMA throughout the development of MLMT, highlighting the importance of developer outreach to regulatory bodies when developing novel outcome measures. The test was also discussed during a workshop organized by patient advocacy groups and the FDA, the goal of which was improving clinical trial design and endpoints for AMD (age-related macular degeneration) and IRD.
Stakeholders’ Acceptance
Patient advocacy groups and developers regularly engage with regulatory authorities in attempts to secure approval of clinical trial designs and novel endpoints, but payers are not commonly involved in such discussions. However, failure to gain acceptance by these stakeholders can result in commercialization challenges.
For example, as highlighted above, publicly available information indicates that Spark Therapeutics engaged repeatedly with the FDA and the EMA, but, according to these documents, payers were not among the stakeholders regularly consulted in the development of the MLMT. When the Institute for Clinical and Economic Review’s (ICER)3 health economists assessed Luxturna’s economic value, they struggled to evaluate the intervention based on the improvement on the MLMT endpoint. As a result, ICER performed its assessment mostly based on visual acuity, a traditional ophthalmic endpoint. ICER’s decision resulted from a lack of data necessary to derive a utility curve based on the MLMT test, which is a critical step in the process of assessing cost effectiveness. Needless to say, ICER deemed Luxturna not cost-effective under most of the scenarios considered. We can speculate that if Spark had engaged in more depth with ICER as well as payers during the development of the MLMT test, it is possible the company could have identified ICER’s need for a MLMT utility curve prior to the evaluation, and ultimately such an approach could have resulted in a more positive cost-effectiveness assessment for Luxturna.
ICER itself appears to recognize the need for developers to include payers in the clinical trial design process and the benefits that could result from early discussions, and it recently announced that the institute is considering offering an “early scientific advice program” that could help manufacturers design clinical trial with more patient-centric outcome metrics.
The Role Of Patient Advocacy Groups
It is important to note that developers are not the only entities that can take charge of developing and/or refining new methods to measure patient-centric outcomes. In fact, patient advocacy groups can also lead this effort. As an example, DRG research indicates that Parent Project Muscular Dystrophy (PPMD), a Duchenne muscular dystrophy (DMD) patient advocacy group, is currently developing a DMD outcome measure tailored to the needs of DMD patients and caregivers. The new outcome, performance of upper limb (PUL), is intended to evaluate a patient’s ability to perform tasks that are important to them, such as lifting a cup to their mouth. Importantly, in the process of developing PUL, PPMD has engaged with the FDA and has even involved payers.
Having a patient advocacy group lead the effort to develop new clinical trial endpoints may indeed simplify the process for developers and, owing to their established relationships with clinicians and regulatory bodies, as well as their connection to patients, help secure wider acceptance of the novel metric; however, it is important to acknowledge that, as nonprofit organizations, most patient advocacy groups require some form of financial support to be able to conduct this work. While collaboration with and financial support of advocacy groups should be managed carefully due to potential conflicts of interest, DRG research conducted with executives from these patient organizations indicates that there are several ways for these organizations to accept manufacturers’ support.
Conclusion
Although development and validation of a patient-centric outcome, which will usually represent a new path to market, is a lengthy process fraught with challenges, persistent developers stand to enjoy long-term commercial gains, as these novel endpoints can be instrumental in proving the efficacy of emerging therapies. Drug developers who pay careful attention to the perspectives of different stakeholders poise themselves to add tremendous value to rare disease therapies: regulatory bodies must be consulted to secure approval; patient advocacy groups and treatment providers must be engaged to secure adoption. Finally, market access gatekeepers must be involved in defining the validity of the outcome measure, thus easing the access burden and facilitating patient and physician demand for novel, effective therapeutics.
Notes:
- NORD (National Organization for Rare Disorders) Summit — Oct. 15 and 16, 2018
- Visual acuity measures how eyes function, but not necessarily how IRD patients function. It’s important to note that Luxturna did not deliver a clinically meaningful result on this metric (as assessed by standard ophthalmology criteria — not tailored to IRD patients).
- ICER (Institute for Clinical and Economic Review) is a U.S.-based watchdog for drugs’ value and cost effectiveness. While this is a nonprofit independent organization, payers often lean on ICER evaluations in their coverage decisions.
About The Authors:
 Claudia Dall’Osso, Ph.D., is a principal business insights analyst on the Infectious, Niche, and Rare Diseases team at Decision Resources Group, specializing in niche and rare indications. Before joining DRG, she held a management and strategy consultant position at Precision Medicine Group, where she worked for clients in the biopharmaceutical, medical, device and diagnostic industries. Dall’Osso completed her master’s in management at Harvard University; she also holds a Ph.D. in medical genetics from Brescia University in Italy and a B.S./M.S. degree in medical biotechnology from the University of Milano in Italy. You can reach her at cdallosso@teamdrg.com or connect with her on LinkedIn.
Claudia Dall’Osso, Ph.D., is a principal business insights analyst on the Infectious, Niche, and Rare Diseases team at Decision Resources Group, specializing in niche and rare indications. Before joining DRG, she held a management and strategy consultant position at Precision Medicine Group, where she worked for clients in the biopharmaceutical, medical, device and diagnostic industries. Dall’Osso completed her master’s in management at Harvard University; she also holds a Ph.D. in medical genetics from Brescia University in Italy and a B.S./M.S. degree in medical biotechnology from the University of Milano in Italy. You can reach her at cdallosso@teamdrg.com or connect with her on LinkedIn.
 Ian Love, Ph.D., is a senior business insights analyst in the Infectious, Niche, and Rare Diseases department at Decision Resources Group. He specializes in atopic dermatitis and a diverse group of rare diseases. He received his doctorate in biomedical sciences from the University of Massachusetts Medical School and his B.S. in cell and molecular biology from Worcester Polytechnic Institute. Prior to joining DRG, Love was an instructor in the Department of Internal Medicine at Virginia Commonwealth University, where his work focused on understanding the regulation of programmed cell death by the tumor suppressor p53.
Ian Love, Ph.D., is a senior business insights analyst in the Infectious, Niche, and Rare Diseases department at Decision Resources Group. He specializes in atopic dermatitis and a diverse group of rare diseases. He received his doctorate in biomedical sciences from the University of Massachusetts Medical School and his B.S. in cell and molecular biology from Worcester Polytechnic Institute. Prior to joining DRG, Love was an instructor in the Department of Internal Medicine at Virginia Commonwealth University, where his work focused on understanding the regulation of programmed cell death by the tumor suppressor p53.