
India Making Way For Separate Clinical Trials Rules
By Bobby George, Ph.D., VP and head of regulatory affairs, Reliance Life Sciences Pvt. Ltd.

The key law that governs the pharmaceutical industry in India is the Drugs and Cosmetics (D&C) Act, 1940 and Rules, 1945. Over time, several amendments have been made to the D&C Act and rules. Schedule Y and Part XA (which covers rules 122A, 122B, 122D, 122DA, 122DAB, 122DAC, 122DB, 122DC, 122DD, and 122E) describe the various procedures for importing or manufacturing new drugs for sale or undertaking clinical trials (CTs) in the country. The Central Drugs Standard Control Organization (CDSCO), the national regulatory agency (also known as the Central Licensing Authority), regulates CTs in India.
In February 2018, the Indian Ministry of Health (MoH) released the draft New Drugs and Clinical Trials Rules after consultation with the Drugs Technical Advisory Board, the country’s highest technical body under the D&C Act. The draft notification, GSR-104(E), with 12 chapters and eight schedules, was posted on the CDSCO website (http://cdsco.nic.in/) for stakeholder’s comments. The MoH is expected to assess objections and suggestions and release the final rules, which would supersede Schedule Y and Part XA. Key updates or changes proposed in GSR-104(E) are discussed in this article.
New Drug Definition
New drugs are currently defined under rule 122-E of the D&C Act. This definition is proposed to be broadened per GSR-104(E) to include a modified or sustained release form of a drug or novel drug delivery system of any drug approved by the Central Licensing Authority (which is the Drug Controller General of India [DCGI] office), living modified organism, monoclonal antibody, stem cell, and gene therapeutic product or xenografts intended to be used as drug. Further, draft GSR-334(E), released on April 4, 2018, defines stem cells and cell-based products as “a drug which has been derived from processed cells including cell or tissue which has been processed by means of substantial or more than minimal manipulation with the objective of propagation and/or differentiation of a cell or tissue, cell activation, and production of a cell line, which includes pharmaceutical or chemical or enzymatic treatment, altering a biological characteristic, combining with a non-cellular component, manipulation by genetic engineering including gene editing and gene modification.” With this, CTs undertaken with these products would legally come under the ambit of CDSCO.
Application Fees
The current and the proposed fees (in India rupee) per draft rules for some applications related to CTs and bioavailability/bioequivalence (BA/BE) studies are summarized in the table below. Apart from an increase in fees by multiple folds, there have been new categories* for which fee-based review has been proposed. For example, there is a proposal to start the process of formal meetings for pre-submissions and post-submissions of the sponsor/applicant with an authorized person from the CDSCO for seeking written guidance about the regulatory requirements and procedures. Currently only informal meetings are held, with no written guidance released by the CDSCO after such meetings.
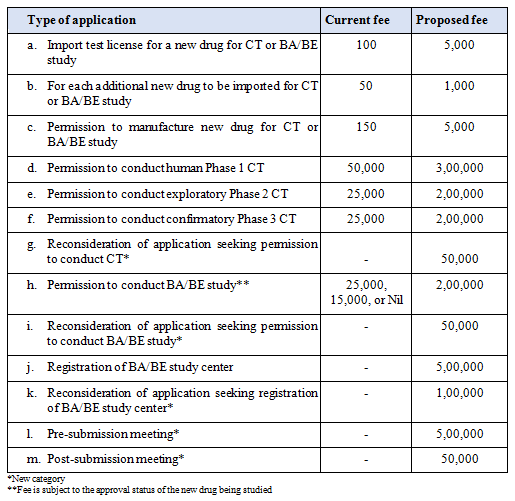
GSR-104(E) also has a provision for complete fee waiver provided the CT is to be conducted by a person of an institution or organization funded or owned, wholly or partially, by the central government or a state government.
While there has been opposition to this spike in fees from some quarters, MoH is of the view the fee hike is long overdue considering the growth achieved by the pharmaceutical sector over the years. Further, to counter the corresponding increase in application load efficiently, the government needs funds and they have to be generated from the industry, on the lines of the user fees charged by the U.S. FDA or by EU agencies. The fee hike is also expected to supplement the associated cost of reviewing submissions and inspecting facilities in a more efficient and effective manner.
Timelines For Review Of Applications
CTs for a new drug can be initiated only after the CDSCO has granted permission and the ethics committees (ECs) of the relevant sites have approved conduct of the study protocol. Presently, there is no mention of specific timelines under the D&C Act for review of different types of new-drug-related submissions. In GSR-104(E), there are defined timelines for the CDSCO to review and CT and BA/BE studies submissions and these would shorten the overall clinical development timelines.
- For CT application with a new drug or investigational new drug which has been discovered, researched, and developed in India, with a proposal to manufacture and market in India: within 45 days of receipt of application. Further, if no communication is received by the applicant from the CDSCO within that period, the permission for conduct of the study would be deemed to have been granted.
- For new drugs already approved and marketed in countries specified by the CDSCO: within 90 days of receipt of application.
- For BA/BE studies of new drugs for export submission: within 90 days of receipt of application.
- If the applicant/sponsor intends to request the CDSCO to reconsider their application for a CT or BA/BE study, they need to apply within 60 days of rejection of application.
Biomedical And Health Research
The Indian Council of Medical Research (ICMR) has “Ethical Guidelines for Biomedical Research on Human Subjects” (released initially in 2000, amended in 2006 and 2017) which addresses general and ethical issues involved in clinical evaluation of drugs/devices/diagnostic vaccines and herbal remedies. The revised guidelines are applicable to all biomedical, social, and behavioral science research for health conducted in India involving human participants, their biological material, and data. Although the guidelines are not yet legislated, they are indirectly mandated through amendment of Indian Medical Council Act, 2002, and Schedule Y amendment of D&C Act in 1995. Having said that, GSR-104(E) has included “Biomedical and health research” and laid out the requirements for carrying out such studies. Such research has been defined to include “studies on basic, applied and operational research or clinical research designed primarily to increase scientific knowledge about diseases and conditions (physical or socio-behavioral); their detection and cause; and evolving strategies for health promotion, prevention, or amelioration of disease and rehabilitation but does not include CT.” Further, ECs reviewing biomedical and health research proposals should register with the authority designated by the Department of Health Research (and not with the CDSCO), within the MoH.
Academic Clinical Trials
GSR-104(E) defines academic trials as “CTs done purely for academic research, whereby the data generated on the drug is not intended for regulatory submission (i.e., if the trials are not for claiming permission of new drug for marketing as per D&C Rules).” Such trials are not required to be submitted to the CDSCO for review and approval and can be approved by the respective ECs. When in doubt, the EC should inform the CDSCO in writing of its views within 30 days of receipt of application. The CDSCO, for its part, should provide necessary clarification in writing within 30 days to the EC, failing which it shall be presumed that no permission from the CDSCO is required. Academic CTs shall be conducted in accordance with the approved CT protocol by the EC, ethical principles specified in guidelines for biomedical research on human participant, and notified by the ICMR.
Clinical Trial Registry
CTs must be registered at the Clinical Trials Registry of India (CTRI) portal (http://ctri.nic.in/) before enrolling the first patient in the study. This was allowed to be done retrospectively until April 1, 2018. Mandatory fields in the database are based on the requirements for registration of the WHO and the International Committee of Medical Journal Editors. Registration is free of charge, and once a CT is registered, it must be updated by the applicant. As part of an agreement on standards framed by the WHO, some of the research institutes and funding agencies (ICMR, PATH, U.K. Medical Research Council, etc.) have agreed to develop and implement policies requiring all CTs they fund, co-fund, sponsor, or support to be registered in a publicly available registry. They have also agreed that all results will be disclosed within specified timelines on the registry or by publication in a scientific journal. GSR-104(E) also proposes BA/BE studies of investigational new drugs be registered with the CTRI before enrolling any subject in the study. “Investigational new drug” is defined in GSR-104(E) as “a new chemical or biological entity or substance that has not been approved for marketing as a drug in any country.”
Compensation And Free Medical Management For CT Subjects
Per the D&C Act, in case of injury to the CT subject, he/she shall be given "free medical management” as long as required or until it is established the injury is not related to the CT, whichever is earlier. If the injury is found to be related to the CT, the subject is entitled to financial compensation over and above any expense incurred on medical management. In the case of CT-related death, the subject’s legal heir/nominee would be entitled to financial compensation. Further, the financial compensation has to be paid only upon receipt of a CDSCO letter, which follows the receipt of opinion from an independent expert committee (constituted by the MoH). Currently, no compensation is to be paid for injury or death of a trial subject if it is proven to be unrelated to the trial.
However, per GSR-104(E), in cases of serious adverse events (SAEs) with death or permanent disability as an outcome of a CT or BA/BE study, the sponsor or their representative shall have to pay an interim compensation (60 percent of the total amount) within 15 days of receipt of the EC opinion. The remaining amount must be paid within 30 days of receipt of a letter from the CDSCO, provided it is confirmed the SAE was related to the study. The interim compensation paid would not be recoverable irrespective of the causality relationship to the study. In case of SAEs other than deaths and permanent disability, the sponsor or its representative shall have to pay the compensation for a CT-related injury within 30 days of receipt of the EC opinion.
Post-Marketing Studies
There used to be confusion among sponsors and CROs on the exact regulatory requirements for carrying out post-marketing studies of new drugs for approved indications. While some firms used to submit only periodic safety update reports (PSURs), others conducted studies, with or without CDSCO approval for their study protocols. GSR-104(E) has clearly differentiated the requirements for carrying out Phase 4 CT and post-marketing surveillance studies for new drugs. The PSUR submission requirements continue to be the same as cited previously in Schedule Y.
i) Phase 4 (post-marketing) trial
These include additional drug-drug interactions, dose-response, or safety studies and trials designed to support use under the approved indications, e.g., mortality/morbidity studies. Such trials need to be conducted under an approved protocol with defined scientific objectives, inclusion and exclusion criteria, safety efficacy assessment criteria, etc. with the new drug under approved conditions for use in an approved patient population. In such trials, the ethical aspects for protection of rights, safety, and well-being of the trial subjects shall be followed per the regulatory provisions, including that for compensation in case of CT-related injury or death and GCP guidelines. In such trials, the study drug may be provided to the trial subject free of cost unless there is specific concern/justification for not doing so, to the satisfaction of the DCGI office and the EC.
ii) Post-marketing surveillance study or observational or non-interventional study for active surveillance
Such studies are conducted with a new drug under approved conditions of its use under a protocol approved by the DCGI office with scientific objective. Inclusion or exclusion of subject is decided per the recommended use according to prescribing information/approved package insert. In such studies, the study drugs are part of the treatment of the patient in the wisdom of the prescriber included in the protocol. The regulatory provisions and guidelines for the CT of a new drug are not applicable in such cases, as the drugs are already approved for marketing.
iii) Post-marketing surveillance (PMS)
Per Schedule Y, PMS is mandatory for four years post-approval of new drugs in the country. The applicants are now mandated to have a pharmacovigilance system in place for reporting to the licensing authority. PMS should include spontaneous/voluntary reporting including reporting from scientific literature, publications, or meetings; voluntary or required reporting from observational studies and randomized CTs; drug-induced injury; detection of events not seen in CTs such as new, previously unknown AE or new drug interactions; and any observed increase either in frequency or severity of a known AE.
Post-Trial Access
Currently, in line with the ICMR guidance, post-trial access arrangements or other care must be described in the study protocol (if not initially, then through an amendment) so the regulatory and EC review may consider such arrangements. Information must also be disclosed to subjects during the informed consent process. GSR-104(E) specifies the requirement for post-trial access of investigational product for needy patients. According to the draft rules in GSR-104(E), the sponsor shall provide post-trial access by giving the drug free of cost to the CT patient, per directions of the CDSCO, and in special circumstances on the recommendations of the investigator and the EC with written consent of the patient or the legal heir of such subject, as the case may be. However, the sponsor shall have no liability for post-trial use of an investigational new drug or new drug.
Archival Of Records And Retention Samples
Schedule Y does not explicitly specify the period for which CT or BA/BE study-related documents must be archived. GSR-104(E) proposes to maintain the data and records for five years after completion of the CT/BA/BE study or at least two years after the expiration date of the batch of the new drug product, whichever is later.
In terms of sample retention in a BA/BE study, Schedule Y has been silent. However, per GSR-104(E), all samples of test and reference drug products used in a BA/BE study should be retained by the organization carrying out the study for five years after the conduct of the study or one year after the expiry of the drug, whichever is later. Each reserve sample should consist of a quantity sufficient to carry out twice all the in vitro and in vivo tests required during a BA/BE study.
Orphan Drug Registration System
Unlike in developed markets, under the present rules, India does not have separate fast-track mechanisms to approve orphan drugs. The waiver of CTs on an Indian population for approval of new drugs that have already been approved outside India can be considered only in cases of national emergency, extreme urgency, an epidemic, drugs for rare diseases, and drugs indicated for conditions/diseases for which there is no therapy. In case a local CT waiver is required for any category other than those above, the matter should be brought before the technical review committee for consideration. However, this option has rarely been exercised for orphan drugs (approved in the U.S. and/or EU), which were allowed to be imported and marketed in India without local CT on an Indian population, subject to the condition to conduct a Phase 4 trial on an Indian population.
GSR-104(E) defines orphan drugs as a “drug intended to treat a condition which affects fewer than 200,000 people in India.” The proposed rule lays down a fast-track approval process and special status for orphan drugs, including:
- no fee shall be chargeable for CT filing
- expedited review
- surrogate end points shall be considered instead of standard outcome measures in CTs
- if remarkable efficacy is observed with a well-defined dose in Phase 2 of a CT, it may be considered for marketing approval
- after accelerated approval, post-marketing trials are to be conducted to validate anticipated clinical benefits.
Some of the above processes/mechanisms may be followed to expedite the development and approval of new drugs intended to be used in life-threatening/serious diseases of special relevance to India as well.
Other Proposed Measures
- There had been instances of firms delaying the initiation of studies even after grant of study approval from the CDSCO. GSR-104(E) mandates firms initiate CTs and BE studies by enrolling the first subject within one year from the date of grant of permission. Firms failing to do so have to seek prior permission from the CDSCO. Further, the permission to conduct the CT shall remain valid for two years from date of issue, with an extension by another two plus one years under exceptional circumstances based on written request to the CDSCO.
- The permission to manufacture and the license to import new drugs for undertaking a CT or BA/BE study shall be issued with a validity of three years, as opposed to one year previously. These could be extended by one year upon written request to the CDSCO.
- Labelling requirements have also been defined for any new drug or investigational new drug manufactured or imported for the purpose of a CT or BA/BE study. No alteration of label shall be permitted without CDSCO permission.
- The approving ECs and the CT site or BA/BE center, as the case may be, shall be located within the same city or within a radius of 50 km to have better oversight.
- Currently, sponsors are bound to notify CDSCO of EC approval prior to initiation of study at the site. The new rules mandate CDSCO to be notified within 15 days of grant of such approvals.
- Additional information proposed to be included in the informed consent document:
- possibility of failure of an investigation product to provide intended therapeutic effect
- in case of a placebo-controlled trial, administration of the placebo shall have no therapeutic effect.
Path Forward
Amendments through GSR-104(E) in the D&C Act intend to regulate CTs, post-marketing studies, and newer areas of science (like biological products, stem cells, and cell-based products), as they cannot be effectively regulated under the present law. Steps being taken by the MoH toward harmonization of standards and convergence of regulatory practices are laudable. Comments from stakeholders will likely result in certain amendments of the provisions under GSR-104(E) rules (such as special considerations regarding biological products like vaccines and r-DNA derived drugs) before they are finalized into law.
Declaration: The author declares no conflict of interest.
About The Author:
 Bobby George, Ph.D., is head of regulatory affairs at Reliance Life Sciences (Navi Mumbai, India). He is responsible for regulatory services across all of the company’s business verticals (pharmaceuticals, biosimilars, plasma proteins, etc.). He has a doctorate in pharmacology and over 20 years of industry experience. He has written 34 publications in peer-reviewed journals and three book chapters. George recently authored and published a book titled “The Act that Wasn’t,” in which he deliberates on the “Acts” (laws) and “acts” (wrong deeds) of those in the drug and healthcare industry, citing several examples of the unholy nexus between stakeholders.
Bobby George, Ph.D., is head of regulatory affairs at Reliance Life Sciences (Navi Mumbai, India). He is responsible for regulatory services across all of the company’s business verticals (pharmaceuticals, biosimilars, plasma proteins, etc.). He has a doctorate in pharmacology and over 20 years of industry experience. He has written 34 publications in peer-reviewed journals and three book chapters. George recently authored and published a book titled “The Act that Wasn’t,” in which he deliberates on the “Acts” (laws) and “acts” (wrong deeds) of those in the drug and healthcare industry, citing several examples of the unholy nexus between stakeholders.