
Key Insights From The 2022 PDA/FDA Joint Regulatory Conference
By Enith Morillo, Cadoret Global Inc.

With over 800 registrants from across 18 countries, the PDA/FDA Joint Regulatory Conference celebrated its 31st anniversary this year. Naturally, being back in person in a post-pandemic era was the most celebratory theme of this conference, which is a confluence of regulators from the FDA and other health authorities, industry leaders, and academia.
In an opening session, Peter Marks, MD, Ph.D., director, CBER, FDA, touched on the challenges faced and lessons learned during the pandemic. Among the biggest challenges encountered were the agility and process knowledge required to expedite product development and manufacturing scale-up, overcoming manufacturing capacity constraints, and streamlining the regulatory pathways for early access to medicinal products, particularly the Covid vaccine.
Among the lessons learned, Dr. Marks remarked, were the best practices for communication between FDA and sponsors, which pivoted from the traditional FDA meeting types to more “live,” informal, and timely interactions aimed to expedite communication and allow for critical decision-making. During this time, the FDA also embraced the use of virtual tools not only for improved communication but for remote assessments, which aligns with the agency’s risk-based approach to surveillance.
The pandemic also put a spotlight on the traditional process for drafting, reviewing, and publishing industry guidances. This resulted in a more balanced and expeditious approach to provide the industry with the necessary tools to immediately execute on some of the challenges brought about by the pandemic.
Global regulatory confluence and additional effort toward mutual recognition significantly increased throughout the pandemic, with regulatory agencies forging new communication pathways for sharing of pharmacovigilance information.
Although the FDA did not necessarily witness a significant increase in drug shortages during the pandemic, industry encountered several bottlenecks in manufacturing medicinal products, such as shortages in raw materials, disposable supplies, and skilled workforce. Some of these challenges continue nowadays and were among the topics of discussion at the recent ChemOutsourcing conference in New Jersey, the largest and longest running API and pharma ingredients conference in the U.S.
If the pandemic taught us anything, it is that speed and quality go hand in hand. This was the center theme of a session presented by Sandra A. Boyd, drug national expert from the FDA’s Office of Regulatory Affairs (ORA). In her session, she illustrated the importance of a quality infrastructure that is designed to prevent fraud by counteracting the rationalization, pressure, and lack of detectability that, combined, may lead to it. When employees in an organization understand the why of what they do, it is easier to avoid the rationalization for cutting corners that eventually leads to falsification of data. Similarly, when they are given adequate resources for the tasks at hand, it is easier for employees to adhere to established procedures and standards and maintain compliance, while having a strong quality culture and presence to ensure that lapses and negative trends are identified and addressed before they compound.
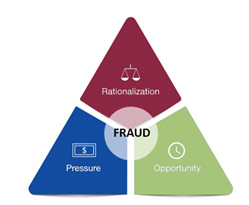
Figure 1 – The Fraud Triangle
Donald D. Ashley, director of the FDA CDER’s Office of Compliance, further delved into this in his session on sustainable compliance. The session emphasized the importance of a quality culture that is endorsed and driven by leadership, with daily decisions on issues involving resources, equipment, materials, maintenance, etc., noticeably influencing the direction of an organization and eventually the quality of the products they supply.
In the session, he illustrated sustainable compliance where a proactive approach leads to a continuous state of control and high-quality products, as compared to compliance efforts that are reactive and aimed at achieving satisfactory inspectional outcomes, which can at times lead to quality and compliance issues.
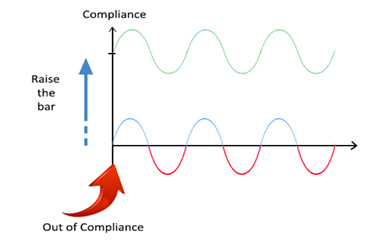
Figure 2 – Sustainable Compliance
Emphasis was placed again on senior management’s critical role to ensure the facility is in an adequate state of repair, the quality unit is enabled and empowered to foster a quality culture, and operations count on adequate resources to meet the daily demands without compromising quality. This was best summarized by Moderna’s recently appointed CEO, Juan Andres, when he stated problems come when management does not know how to make medicines. It is, therefore, no surprise that warning letters are generally addressed to senior leadership.
The conference was packed with engaging sessions on a variety of topics, including sessions led by PDA’s interest groups (IGs), such as one on data integrity. With a panel that included Carmelo Rosa, division director of FDA’s Office of Manufacturing and Product Quality, CDER, among others, this session drew parallels between GMP and data integrity. Unlike GMP, where phase-appropriateness is critical throughout the development life cycle and the quality system’s maturity increases as the product moves through clinical phases and into market, data integrity either exists or doesn’t.
Indeed, data integrity must be present at all stages of development, with its accuracy and reliability being required and preserved. The expectation is that any surfacing issues associated with data integrity are proactively prevented and adequately investigated when they occur, and that industry is transparent with regulatory agencies when concerns arise that need to be addressed.
For the agency, surveillance continues to be risk-based, with inspections being only a part of a multi prong approach. Judy McMeekin, Pharm.D., associate commissioner for regulatory affairs, held a session on updates from the FDA’s ORA. One of the topics she touched on is ORA’s plans to launch a pilot program on foreign unannounced and for cause inspections. To date, foreign inspections are generally announced; through this program, the agency aims to determine if inspectional outcomes of foreign sites are impacted by these being announced.
She also touched on the different types of regulatory remote assessment tools and the FDA’s collaboration with other health authorities globally, with potential expansion of the mutual recognition agreement to cover veterinary products.
The session was complementary to one presented by Susan Laska, M.S., on the Pharmaceutical Inspection Co-Operation Scheme (PIC/S) remote assessments. Her session covered the recent collaborative efforts needed across global regulatory agencies and industry groups to pave the way for increased mutual recognition. In addition, the session touched on the differences between desktop audits, fully or partially interactive remote assessments, and hybrid inspections, and highlighted the importance and ongoing focus of their working group to standardize terminology and best practices to ensure alignment and maintain a risk-based approach to both supplier management and regulatory assessments. Interestingly, the crucial role of IT was also brought to light, as oftentimes technical issues can surface during virtual sessions that hinder the effectiveness of remote assessments.
Overall, the conference captured what we, as industry, regulators, and academia, have learned from the pandemic and reinforced the importance of sharing knowledge, best practices, and collaboration to reach a common goal: public health.
About The Author:
 Enith Morillo, M.Sc., is the founder, president, and principal consultant of Cadoret Global, a quality and compliance consulting firm that specializes in supporting virtual, early-stage, and small pharmaceuticals in taking their investigational drug through development and into Phase 1-2 clinical trials. Cadoret Global is a woman-owned and minority-owned certified company and an alumni of the Goldman Sachs 10,000 small businesses program. She can be reached at Enith.Morillo@CadoretGlobal.com or on LinkedIn.
Enith Morillo, M.Sc., is the founder, president, and principal consultant of Cadoret Global, a quality and compliance consulting firm that specializes in supporting virtual, early-stage, and small pharmaceuticals in taking their investigational drug through development and into Phase 1-2 clinical trials. Cadoret Global is a woman-owned and minority-owned certified company and an alumni of the Goldman Sachs 10,000 small businesses program. She can be reached at Enith.Morillo@CadoretGlobal.com or on LinkedIn.