
Navigating China's Biologics Approval And Accelerated Pathways
By April Wang, Accestra Consulting Company

China's pharmaceutical market, the second largest in the world after the United States, is a hotspot for international pharmaceutical companies. As they seek to tap into this vast market, understanding the regulatory landscape and approval pathways is crucial. In this article, we will delve into the standard biologics approval procedures in China and explore accelerated pathways that can expedite the entry of certain products into the country.
Understanding China's Regulatory Framework
The regulatory framework for pharmaceuticals in China is a multi-tiered structure, with various government departments playing essential roles. At the top are the Congress and the State Council, responsible for drafting laws that form the foundation of the regulatory system. The National Medical Products Administration (NMPA), formerly known as the CFDA, is the second tier, overseeing market approval and post-marketing surveillance. Below NMPA is the Center for Drug Evaluation (CDE), responsible for drafting technical guidelines and reviewing application dossiers. Two other important departments under NMPA are the National Institutes for Food and Drug Control (NIFDC) and the Center for Food and Drug Inspection (CFDI), responsible for quality testing and on-site inspections, respectively.

Biologics In China: A Closer Look
In China, drug registration is divided into three primary categories: chemical drugs, biological products, and traditional Chinese medicines. Biologics are further categorized into three groups based on their functions: preventive biologics, therapeutic biologics, and in-vitro diagnostic biologics. For the purpose of this article, we will focus on therapeutic biologics.
The registration procedure includes various stages such as Investigational New Drug application (IND)/Clinical Trial Application (CTA), New Drug Application (NDA)/Biologic License Application (BLA)/Marketing Authorization Application (MAA), license renewal, and variation application. Three review pathways are available: standard review, accelerated review, and special pathways. We will further discuss the key milestones and pathways in the following sections.
Navigating The Standard Registration Pathway Procedures: Two Key Milestones
1. Clinical Trial Application (CTA)
- Step 1: CDE communication meeting
It is recommended, especially for innovative drugs, to apply for a communication meeting, also known as pre-IND meeting, with CDE to discuss the critical R&D issues that are not covered by regulations or technical guidelines before clinical application, including the possibility of clinical trials exemption. In addition to a written response, online or face-to-face meetings would be arranged if necessary.
- Step 2: CTA dossier submission
The sponsor submits the application in ICH M4 CTD format, which undergoes a five-working day format acceptance review. Up to this point, the eCTD format is not mandatory. The CDE requires all applications to be submitted electronically, replacing the hardcopy submission.
- Step 3: CDE technical review and approval by default
CDE conducts a 60-working day technical review and if the materials are comprehensive and convincing, the application will be approved by default.
- Step 4: Other application activities before launching clinical trial
The sponsor needs two to three months to complete Ethical Approval, Human Genetic Resource Administration notification, as well as register on the clinical trials platform.
To complete all the necessary steps, it usually takes six to 10 months in total to initiate a clinical trial in China.
2. Marketing Authorization Application (MAA)
- Step 1: Registration dossier submission
Once all documents for the marketing authorization application in China are ready, applicants are required to prepare the registration dossier in both Chinese and the language of origin, following the ICH M4 CTD format, and then submit it to CDE.
- Step 2: CDE format compliance and material completeness check
CDE checks the format compliance and material completeness within five working days and, if necessary, requests supplementary documents to be submitted within 30 working days. Sample testing for registration and administration fee payment will be required immediately upon the acceptance of application.
- Step 3: CDE first round technical review
After a specified queuing period, CDE initiates the technical review process in two phases: an initial review lasting 40 working days followed by a comprehensive review of the entire product dossier, which takes up to 160 working days. During this initial technical assessment, CDE evaluates whether a GMP/GCP/GLP inspection is required, based on the risk assessment of the imported drug. Inspections can be conducted either remotely or on-site. Notably, the on-site registration inspection is not mandated to occur prior to the drug's marketing authorization approval; it can also be conducted post-approval.
- Step 4: Deficiency letter and second round technical review
Following the initial technical review, if additional documents are required, CDE will issue a deficiency letter (a request letter for supplementary documents), thereby extending the technical review period by an additional one-third of the original review duration.
- Step 5: CDE conclusion and NMPA administrative result
CDE summarizes the review results with a conclusion. For positive results, NMPA grants approval for the product and issues a drug registration license.

The entire process usually takes around 14 to 20 months, but this may vary depending on the specific situation.
Accelerated Review Pathways
The accelerated review policies in China consist of two distinct parts. This approach represents a significant draw for many innovative drug companies seeking expedited market entry for their products:
The first part is list-based management: Since 2018, NMPA has introduced three batches of innovative drugs in urgent clinical need with overseas approval. Products on the lists can obtain approval in China within three to six months, excluding sample testing time. They may also be exempt from the requirement for Chinese ethnic clinical data, and conditional approval is possible even with insufficient Chinese population data.
The second part is the accelerated policy system. Four key pathways are outlined in the table below, according to the new “Provisions of Drug Registration” in 2020:
| Pathways | Prerequisites |
|---|---|
| 1. Breakthrough Therapy Drug | √ New drugs that have not been approved in any country (Innovative drugs or modified new drugs). √ Adequate evidence showing clear clinical advantages. √ For diseases that pose a severe threat to life or significantly diminish the quality of life, lack effective prevention and treatment options, or offer clear advantages over existing treatment methods |
| 2. Conditional Approval |
a. Drugs for treating severe life-threatening diseases with no effective treatment available, where clinical trials of the drug have already demonstrated efficacy and can predict its clinical value; c. Vaccines urgently required to address significant public health emergencies, or other vaccines as designated by the NMPA, where the assessed benefits significantly outweigh the risks. |
| 3. Priority Review |
a. Drugs in critical clinical shortage (NHC catalogue). b. Innovative new drugs and improved new drugs for the prevention and treatment of serious infectious diseases and rare diseases (NHC catalogue). c. New varieties, dosage forms, and strengths of pediatric drugs that are compatible with children's physiological characteristics. d. Vaccines urgently needed for disease prevention and control, and innovative vaccines. e. Drugs included in breakthrough therapy program. f. Drugs eligible for conditional approval. g. Other circumstances determined by NMPA. |
| 4. Special Approval | Drugs that address public health emergency defined by NMPA according to the law, such as COVID-19 pandemic. |
The differences and benefits of these pathways are summarized in the table below:
| Pathways | Breakthrough Therapy | Conditional Approval | Priority Review | Special Approval |
|---|---|---|---|---|
| Submission Time | • During clinical trial, usually before Phase III clinical trial. | • CDE communication during clinical trial and before MAA. • Submit formal application when applying for MAA. |
• CDE communication before MAA. • Submit formal application when ready for MAA. |
• CDE communication before CTA/MAA. • Submit formal application when applying for CTA/MAA. |
| Benefits | • CDE meeting support (Type 1 meeting with shorter waiting time). • Ongoing communication and guidance from CDE. • Priority review. |
• CDE meeting support (Type 1 or 2 meeting). • Priority review. • Approval based on surrogate endpoints, interim data, or early clinical trial data. |
• Shortened review timeline. • Priority in arranging onsite inspection, sample testing, and generic name review. • Rolling submission of materials upon an agreement with CDE. |
• Extensive CDE support and guidance from early stage. • Significant reduction in both review and approval timeline. |
| Review Timeline | • Eligibility granting: 45 workdays review + 5 workdays publicity. • MAA review: 200 workdays → 130/70 workdays. |
• Apply for eligibility together with MAA. • MAA review: 200 workdays → 130/70 workdays. |
• Eligibility granting: 5 workdays review + 5 workdays publicity. • MAA review: 200 workdays → 130/70 workdays. |
• Format check: 5 workdays → 24 hours. • CTA/MAA review: 60/200 workdays → 15 workdays. • Approval: 20 workdays → 3 workdays. |
Moreover, in March 2023, the CDE unveiled a new guideline designed to accelerate the review process for breakthrough therapy drugs, pediatric innovative drugs, and innovative orphan drugs. This all-encompassing guideline amalgamates multiple preferential policies into a unified framework.
Special Policies To Access China
Apart from nationwide policies, three noteworthy regulations offer special pathways to regional/designated hospitals as shown in the table below:
| Regulation | Competent Authority | First Issuance Time | Scope of Urgent Clinical Import Drugs | Review Time |
|---|---|---|---|---|
| 1. GBA* Urgent Clinical Use Drug Policy | Guangdong Provincial MPA | Nov. 2020 | • Urgently needed for clinical use by designated medical institutions. • Approved in Hong Kong or Macao (faster and easier approval processes). • Non-psychotropic or other restricted drugs. |
• Around 2 months of review time. • 1 year validation. |
| 2. Hainan Bo'ao International Medical Tourism Pilot Zone Policy | Hainan Provincial MPA | Dec. 2018 | • Urgently needed for clinical use by designated medical institutions. • Approved in U.S., EU, Japan, or other countries or regions. • Non-psychotropic or other restricted drugs. |
5-7 workdays of review time. |
| 3. NHC & NMPA Urgent Clinical Use Drug Policy | National Health Commission and NMPA | June 2022 | • Urgently needed for clinical use. • Not available in China but approved overseas. • Drugs either for rare diseases or to prevent/treat life-threatening diseases without effective/obvious clinical advantage treatment. |
NMPA shall make an approval reply within 3 workdays after receiving the written feedback from NHC. |
*GBA stands for Guangdong-Hong Kong-Macao Greater Bay Area
All three policies allow urgently needed drugs that have been approved in specific regions to be directly imported and used in designated hospitals in China without a registration requirement.
Optimizing Registration Strategy
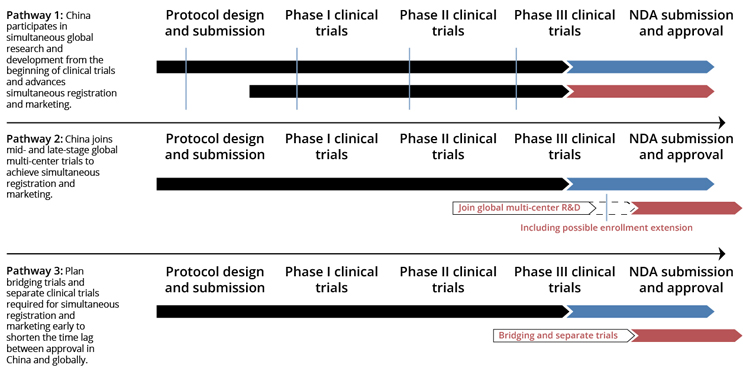
Source: RDPAC, Series Report 4: Promoting Simultaneous R&D, Registration and Review of Innovative Drugs, 2021
In formulating a registration strategy for innovative drugs in China, there are three key options to consider. In the past, pathway 3 was the most adopted, initiating the registration process in China post the completion of Phase 3 clinical trials or approval in other countries. Nowadays, pathway 2 is becoming an alternative to involve China earlier in the process by incorporating it into multi-regional clinical trials (MRCTs) from Phase 2 or 3. Pathway 1 is a more proactive strategy encouraged by NMPA, which involves preparing for China registration right from the outset. The selection among these options should be carefully tailored to the product's characteristics, development stage, and the regulatory support resources in China. Each approach presents its advantages and challenges, significantly influencing the overall clinical trial strategy.
When contemplating the integration of overseas clinical data into China, two critical factors must be taken into account:
- First, what are the development and registration stages of the product, considering whether it is already available in the overseas market or still undergoing R&D?
- The second factor is the indication area, determining whether it falls into the category of orphan drugs, pediatric drugs, or other urgently needed drugs in China.
These factors give rise to diverse scenarios that ultimately guide decisions regarding the requisite clinical trials in China. For instance, if the product does not qualify for accelerated review pathways and falls under the standard review process, an evaluation of the indication background, clinical practices, and the availability of MRCT resources in China becomes essential in this decision-making process.
Practical Tips
The following are five key tips for a smooth registration process:
- Identify the registration classification carefully.
- Pay attention to details when designing clinical protocols in China.
- Maintain open communication with CDE.
- Ensure compliance with Chinese pharmacopeia requirements.
- Stay updated on evolving Chinese regulations and practices.
Navigating the biologics approval process and accelerated pathways in China is complex but essential for international pharmaceutical companies seeking access to this dynamic market. Understanding the regulatory landscape and staying informed about the latest policies and guidelines is crucial for a successful entry into the second-largest pharmaceutical market in the world.
About The Author:
 April Wang is a specialist in Chinese regulatory affairs at Accestra Consulting where she helps clients better understand Chinese regulations and supports them through medical product registrations. Contact her at info@accestra.com.
April Wang is a specialist in Chinese regulatory affairs at Accestra Consulting where she helps clients better understand Chinese regulations and supports them through medical product registrations. Contact her at info@accestra.com.