
Navigating FDA's New AI Systems: Practical Tips For Regulatory Success
By Kimberly Chew, Esq., Odette Hauke, and Kathleen Snyder, Esq.

In part one of this series (read the article here), we examined the legal and regulatory implications of the FDA’s adoption of AI tools such as Elsa, focusing on how these advances affect data confidentiality, trade secrets, prompt security, due process, and the challenges of novel modalities. With this foundation established, part two shifts from legal risks to practical strategies, offering actionable guidance for sponsors navigating AI-enabled regulatory review.
This two-part series aims to provide a practical, solution-focused guide to help sponsors realize efficiency gains while designing for legal robustness.
Part one addressed:
- Data confidentiality & trade secrets
- Security of prompts and exchanges
- Due process & transparency
- Operational readiness
- Novel modalities
Part two will address:
- Clarity and completeness in AI-era submissions: How to structure and document filings to preempt AI-driven queries and reduce review delays
- Writing for both human and AI reviewers: Tips for harmonizing narrative and structured data to avoid misinterpretation by automated systems
- Proactive engagement with the FDA: Strategies for early communication to clarify how AI will be used in your review and set expectations
- Trade secret and data protection in practice: Applying minimum necessary disclosure, redaction, and CCI tagging in the context of AI-assisted review
- Meticulous recordkeeping and traceability: Best practices for documenting all AI-related queries, responses, and supporting data for a defensible administrative record
- Identifying and addressing AI limitations: Approaches for detecting and responding to AI errors, hallucinations, or modality-inapt queries while ensuring human oversight
- Building internal AI literacy: Training regulatory and legal teams to recognize and respond effectively to AI-generated content and stay current with evolving FDA guidance
With these practical tools and recommendations, sponsors can maximize the benefits of AI-enabled review while protecting proprietary interests and ensuring regulatory compliance.
Clarity and Completeness in Submission Documentation
Front-load clarity and completeness. With AI-driven reviews, especially those conducted by agentic AI models like Elsa, it is likely that there will be less opportunity for iterative clarification with the reviewer. To avoid delays related to AI-flagged inconsistencies, sponsors should provide clear, well-organized data, prospectively address potential areas of inconsistencies that may cause concern, and anticipate likely questions and supplement their initial filings with data that is likely responsive to such questions. Sponsors could consider conducting internal AI-readiness reviews to simulate how an FDA algorithm might parse the submission to identify ambiguities or gaps before filing. Identifying and fixing any inconsistencies before submission will help streamline the process and avoid extended delays.
Write for both humans and machines. Sponsors should consider including both a narrative that interprets the findings for a human reviewer and structured data that supports the narrative for the AI review. Before submission, confirm that there are no inconsistencies between the narrative and the structured data. AI tools like Elsa take in and repackage information rather than independently interpreting meaning. Ambiguous or vague language can lead to misclassification or misinterpretation. Elsa’s algorithms may detect patterns or anomalies that human reviewers might overlook, leading to new types of information requests or queries that are more data-driven or granular. Adapt the submission formats to better support AI parsing. Use precise terminology, explicit statements, and structured formats (tables, metadata tags) to ensure clarity. This is especially important for novel or complex products where the FDA’s AI tools may not have a standard to compare to.
Proactive Engagement And Early Communication
Engage early with the FDA. Initiate discussions with the FDA to understand how Elsa and other AI tools will be used to review the submission. Early engagement with the FDA helps clarify expectations and allows sponsors to address concerns regarding AI limitations before submission.
Reference statutory and regulatory protections. As discussed above, in all communications and submissions, reinforce expectations regarding confidentiality by citing FDA’s obligations under 21 CFR 314.430, 21 CFR 20.61, FOIA, and FD&C Act protections, and tagging data as CCI.
Trade Secret And Data Protection Strategies
Minimize necessary disclosure. Sponsors should carefully evaluate the minimum necessary disclosure to satisfy regulatory requirements while protecting proprietary information. Consider redaction or anonymization strategies and flag sensitive data for special handling. As mentioned above, reference the FDA’s confidentiality obligations and mark all CCI data as such.
Meticulous Recordkeeping And Traceability
Document everything. Sponsors should maintain detailed records of all data submitted to the FDA and every query/response exchanged with an AI tool. Keep records of both the data provided and the exact queries or outputs generated by the AI. Where the rationale for an FDA request is unclear, sponsors should request clarification in writing to ensure the basis for the information request is understood and can be addressed thoroughly. Sponsors should also maintain timestamps to establish a clear administrative record.
Explicitly reference supporting data. When responding to an FDA request that appears to be an AI-generated finding, the sponsor should cite the specific data or sections of the submission that address the AI’s concerns. Where appropriate, explain why certain AI-flagged issues may not be relevant or material.

Vigilance For AI Limitations And Procedural Safeguards
Identify and flag AI limitations. Actively monitor for gaps, opacities, or hallucinations in the FDA’s outputs. Early reviews of Elsa were concerned with hallucinations, and the FDA is aware of the concerns. Recently, the FDA stated that “the AI tools do not make regulatory decisions or replace human judgement.”1 However, sponsors should be prepared to raise procedural concerns or request clarification and human review when the suspected AI’s reasoning or requests are unclear.
Human oversight remains essential. While AI tools can accelerate review cycles, final fact-checking and decision-making still require human judgment. Sponsors should work with their FDA contact to understand how to escalate concerns.
Building Internal AI Literacy
Develop AI literacy within teams that interface with the FDA. Regulatory and legal teams should be trained to recognize AI-generated content and its limitations. Understanding how AI models operate, their risk profiles, and their documentation requirements is crucial for effective engagement. It is also important for internal teams to have access to AI tools for pre-submission analysis and editing. Identifying and addressing inconsistencies before formal submission will save time in the overall review timeframe.
Stay current with FDA guidance. The FDA has issued draft guidance and frameworks for assessing the credibility of AI models used in regulatory submissions.2 Sponsors should be familiar with these documents and integrate their recommendations into their processes.
Conclusion
The FDA’s commitment to agentic AI tools, including Elsa, marks a transformative step in the FDA’s modernization of scientific review. For sponsors, this evolution offers both opportunity and obligation: accelerated analyses and earlier insights paired with new responsibilities for documentation, confidentiality, and procedural clarity. AI-assisted review will likely become a permanent feature of the regulatory landscape. To navigate it effectively, sponsors must pair technological adaptability with legal rigor, fortifying trade secret protections, maintaining a transparent administrative record, and building organization‑wide AI literacy. By anticipating these shifts, life sciences companies can not only safeguard proprietary interests but also contribute to shaping ethical, efficient, and accountable AI use within the FDA’s review ecosystem.
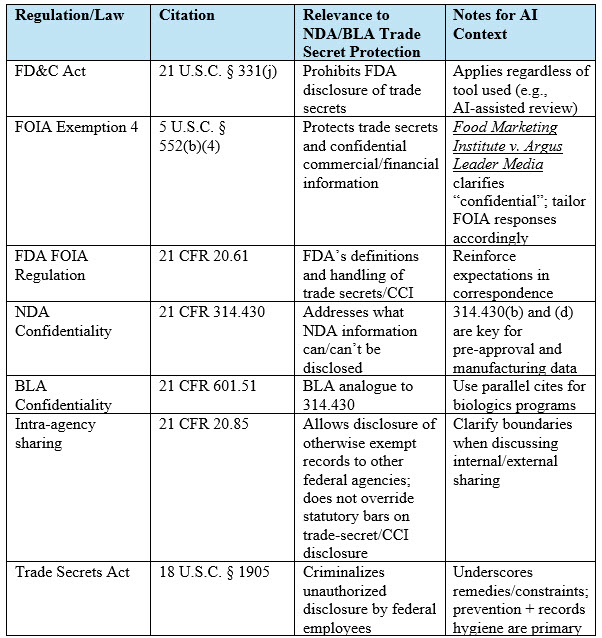
Table 1. Summary of Key Laws and Regulations Protecting NDA/BLA Trade Secrets:
Endnotes:
- https://www.statnews.com/2025/12/01/fda-announces-agentic-ai-elsa-pre-merket-review/#:~:text=The%20agency%20touts%20in%20a%20release%20that,surveillance%2C%20
inspections%20and%20compliance%20and%20administrative%20functions.%E2%80%9D - See U.S. Food & Drug Admin., Press Release, FDA Proposes Framework to Advance Credibility of AI Models Used in Drug and Biological Product Submissions (May 20, 2024), https://www.fda.gov/news-events/press-announcements/fda-proposes-framework-advance-credibility-ai-models-used-drug-and-biological-product-submissions. See also U.S. Food & Drug Admin., Draft Guidance, Considerations for the Use of Artificial Intelligence to Support Regulatory Decision-Making for Drug and Biological Products (May 2024), https://www.fda.gov/regulatory-information/search-fda-guidance-documents/considerations-use-artificial-intelligence-support-regulatory-decision-making-drug-and-biological. See also U.S. Food & Drug Admin., Guidance for Industry and FDA Staff, Artificial Intelligence (AI)-Enabled Device Software Functions: Lifecycle Management and Marketing (Jan. 2024), https://www.fda.gov/regulatory-information/search-fda-guidance-documents/artificial-intelligence-enabled-device-software-functions-lifecycle-management-and-marketing.
About The Authors:
 Kimberly Chew is senior counsel in Husch Blackwell LLP’s virtual office, The Link. Chew is a seasoned professional with a rich background in biotech research, leveraging her extensive experience to guide clients through the intricate landscape of clinical trials, FDA regulations, and academic research compliance. As the co-founder and co-lead of the firm’s psychedelic and emerging therapies practice group, Kimberly is particularly inspired by the potential of psychedelic therapeutics to address mental health conditions like PTSD. Her practice encompasses regulatory due diligence and intellectual property enforcement, particularly in patent infringement and validity.
Kimberly Chew is senior counsel in Husch Blackwell LLP’s virtual office, The Link. Chew is a seasoned professional with a rich background in biotech research, leveraging her extensive experience to guide clients through the intricate landscape of clinical trials, FDA regulations, and academic research compliance. As the co-founder and co-lead of the firm’s psychedelic and emerging therapies practice group, Kimberly is particularly inspired by the potential of psychedelic therapeutics to address mental health conditions like PTSD. Her practice encompasses regulatory due diligence and intellectual property enforcement, particularly in patent infringement and validity.
 Odette Hauke is a global regulatory affairs consultant supporting regulatory strategy across clinical development and registration, with an emphasis on clear regulatory narratives and submission strategies that meet heightened evidentiary expectations. She has 12+ years’ experience directing IND/CTA/NDA/BLA/MAA work across the U.S., EU, UK, Japan, Canada, APAC, and Latin America, and is experienced in integrating AI/ML-enabled regulatory intelligence into decision-making. Previously, she served as associate director of regulatory affairs at AtaiBeckley, leading global regulatory strategy for first-in-class psychedelic and neuropsychiatric programs including VLS-01 (DMT) and EMP-01 (MDMA), navigating novel endpoints, complex trial operations, Schedule I requirements, and evolving global guidance. Earlier, at Memorial Sloan Kettering Cancer Center, she managed 200+ oncology IND submissions and maintained regulatory documentation for 30+ clinical trials, including pediatric research. She holds an M.S. in Regulatory Affairs and a B.S. in Epidemiology.
Odette Hauke is a global regulatory affairs consultant supporting regulatory strategy across clinical development and registration, with an emphasis on clear regulatory narratives and submission strategies that meet heightened evidentiary expectations. She has 12+ years’ experience directing IND/CTA/NDA/BLA/MAA work across the U.S., EU, UK, Japan, Canada, APAC, and Latin America, and is experienced in integrating AI/ML-enabled regulatory intelligence into decision-making. Previously, she served as associate director of regulatory affairs at AtaiBeckley, leading global regulatory strategy for first-in-class psychedelic and neuropsychiatric programs including VLS-01 (DMT) and EMP-01 (MDMA), navigating novel endpoints, complex trial operations, Schedule I requirements, and evolving global guidance. Earlier, at Memorial Sloan Kettering Cancer Center, she managed 200+ oncology IND submissions and maintained regulatory documentation for 30+ clinical trials, including pediatric research. She holds an M.S. in Regulatory Affairs and a B.S. in Epidemiology.
 Based in Boston, Kathleen Snyder practices at the intersection of healthcare and technology, providing clients with practical legal advice on AI governance, strategic technology and commercial contracts, data strategies, intellectual property, and regulatory interpretation. With 20+ years of experience in the healthcare industry, Kathleen has an intrinsic understanding of the healthcare landscape. Her technology-focused transactional practice, coupled with her regulatory experience, gives her a unique perspective that allows her to provide holistic legal advice.
Based in Boston, Kathleen Snyder practices at the intersection of healthcare and technology, providing clients with practical legal advice on AI governance, strategic technology and commercial contracts, data strategies, intellectual property, and regulatory interpretation. With 20+ years of experience in the healthcare industry, Kathleen has an intrinsic understanding of the healthcare landscape. Her technology-focused transactional practice, coupled with her regulatory experience, gives her a unique perspective that allows her to provide holistic legal advice.