
Navigating The Changes To The EU Clinical Trials Regulations 536/2014

The processes for applying for and authorizing clinical trials in Europe have, traditionally, held numerous inefficiencies. Currently governed under the Clinical Trial Directive 2001/20/EC, trial sponsors have been required to submit separate applications for each member state in the European Economic Area (EEA) where they planned to conduct a study. These applications have then been individually assessed by each member state with no collaboration with any other countries involved in the study. Inefficient processes like these have delayed the opening of many clinical trials in Europe, increased the costs of conducting trials, and resulted in divergent decisions across member states that put the development of new therapies in peril.
Fortunately, the way clinical trials are run across Europe is undergoing a major change for the better. The introduction of the EU Clinical Trial Regulation (CTR) 536/2014, that took effect on 31 January 2022, is meant to harmonise the submission and assessment processes, improve cooperation and transparency between member states, and heighten safety standards. This will be achieved through the introduction of a new Clinical Trial Information System (CTIS); a single-entry point platform for the application, authorisation, and supervision all clinical trials conducted throughout Europe. The CTIS simplifies the application and authorisation processes by requiring:
- A single-entry point for all clinical trials applications
- A single set of documents to be submitted for all Member States Concerned (MSC)
- A single fee per member state
- A single authorisation procedure for all clinical trials with strict timelines
- A single member state (Reporting Member State) leading coordination assessment of clinical trial (CT) applications
- A single decision and report
In preparation for the launch of the CTIS GO-LIVE on the 31st of January, it is critical that sponsors understand the requirements for submitting and managing a clinical trial application in the new CTIS. Below THREAD has provided guidance to help interpret the requirements for a clinical trial application and authorisation under the new CTR.
Walking Through the Procedure for Clinical Trial Application & Authorisation in the CTIS
First, the sponsor must submit a single dossier via the CTIS to each MSC where it intends to conduct the trial.
Selection of the Reporting Member State (RMS)
In the application dossier, the sponsor will be required to nominate one MSC to be the Reporting Member State (RMS). The RMS will be responsible for conducting the primary assessment of the Part 1 submission and compiling the assessment report for circulation to the other MSCs. The RMS designation is handled within the CTIS, which tracks which MSC is nominated to be the RMS, whether or not they accept that nomination, and whether or not any other MSCs volunteer to serve as the RMS. All of these details are shared via the CTIS with all MSCs. If more than one MSC expresses willingness, the RMS is chosen by a joint decision by all the MSC. If there is no agreement among MSCs, the RMS role is assigned by the sponsor to their choice of MSC. Once the dossier is submitted into the CTIS it is accessible by all the MSCs, including the RMS.
Requirements for the Clinical Trial Dossier
Under the Clinical Trial Regulation, the application dossier is then divided into two parts:
Part 1 consists of the scientific documentation related to the trial, detailing:
- The anticipated therapeutic intent and benefit for public health
- Potential risks for the subject
- Compliance with manufacturing, import, and labelling requirements
- Additional support documentation provided in the application dossier (See Table 1)
- Part 1 requires a coordinated review by the RMS and all MSCs.
Part 2 will contain the national and participant-level documents and will require assessment by each MSC for its territory (See Table 1)

The sponsor can submit a partial application dossier including only Part 1 to initiate the evaluation process. However, the sponsor must submit Part 2 for all MSCs within two years following notification of the conclusion of Part 1, otherwise the application will lapse.
Navigating the Authorisation Process
Following submission of the dossier, the authorisation process is made up of three distinct phases: Validation, Assessment of Parts 1 and 2 of the dossier, and Final decision. Table 2
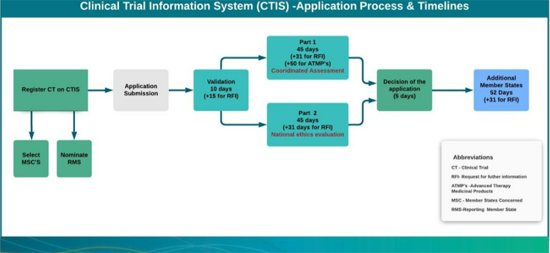
Validation Period (10 Days; up to 15 Days for requests for additional information)
Within 10 days of the submission of the application, the RMS will validate the application for completeness, verify that it falls under the scope of the clinical trial regulation and consider any concerns expressed by the various MSCs. The RMS may send requests for additional information (RFIs) to the sponsor, adding an additional 15 days to the validation process (10 days for sponsor response, five days for RMS decision). If the sponsor fails to respond within the fixed deadline, the application will be withdrawn automatically in all MSCs. If the RMS does not provide feedback within the defined time frame, this will be considered as a tacit validation of the application.
Part 1 Assessment Period (45 day + up to 31 Days for RFI)
Within 45 days from the date the application is validated, the RMS, in collaboration with the MSCs, will perform a review of Part 1 of the application assessment and draft a Part 1 assessment report.
The 45-day assessment period is divided into three parts with defined timelines:
- 26 days - Assessment and draft of Part 1 assessment report by RMS
- 12 days - All MSCs will complete a coordinated review of the draft Part 1 assessment report
- 7 days - Part 1 assessment report is finalised and submitted to sponsor and all MSCs
All this said, the RMS may send a RFI to the sponsor after the 45-day period. In these cases, an additional 31 days may be added to the assessment period (12 days for sponsor response, 12 days for joint review by the MSCs, 7 days for finalisation of the assessment report by the RMS).
If the sponsor fails to respond in 12 days with the RFI response, it will be considered a withdrawal of the application for all MSCs.
Part 2 Assessment Period (45 day + up to 31 Days for RFI)
The Part 2 assessment will be conducted by each MSC participating in the trial for its own country and will involve review by an ethics committee of any regulatory aspects of the clinical trial application that are of a more local/national nature. If Part 2 is submitted at the same time as Part 1, both will be assessed in parallel. The assessment period of Part 2 will contain the same timelines as Part 1 with up to 31 days for RFI and the sponsor must respond within the timelines, or the trial will be automatically rejected.
Each MSC will notify the sponsor through the CTIS as to whether the trial is authorised, authorised subject to conditions, or rejected.
Final Decision (5 Days)
Once the review has been completed by the RMS and all MSCs, the RMS will forward Part 1 assessment reports with a conclusion to whether the conduct of the trial is authorised, authorised subject to conditions, or refused. The final assessment report will be made publicly available on the CTIS.
Adding New MSCs Post-Authorisation
After the authorisation of the initial clinical Trial application, it is possible to add additional MSCs to the trial. This however is only possible if:
-
All original MSCs have notified a decision
-
There is no ongoing assessment of any Part 1 or Part 2 substantial modification procedures in any MSC.
The sponsor will be notified by the MSCs of the national single decision within 52 days of the submission of the application dossier. The new MSC can discuss the application with the RMS and other MSCs within the defined timelines. In the case of RFI, only the RMS may send additional RFI to the sponsor. In this case, the maximum timeline extension becomes 31 days. Additional MSCs can disagree with the conclusion of the RMS Part 1 assessment report as in an initial application, but this does not change the completed Part 1 assessment.
For more information on the new EU regulation 536/2014 and how to implement these changes in your clinical trials, visit THREADresearch.com or contact your THREAD representative.