
Taking Control Of Quality Tolerance Limits In Clinical Trials
By Steve Whittaker, The Avoca Group

The International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) E6(R2) regulations have created a buzz within the industry regarding newly required expectations for quality tolerance limits (QTLs) when conducting good clinical practice (GCP) clinical trials. QTLs have historically been required for good manufacturing practice (GMP) activities, inferring limits at which significant actions must be taken to ensure the manufactured product achieves quality and usability limits.
With regulators now using the same terminology for GCP clinical studies, what is truly expected? ICH E6(R2) describes in Section 5.0.4: “The Sponsor should decide which risks to reduce and/or which risks to accept. The approach used to reduce risk to an acceptable level should be proportionate to the significance of the risk...Predefined quality tolerance limits should be established, taking into consideration the medical and statistical characteristics of the variables as well as the statistical design of the trial, to identify systematic issues that can impact subject safety or reliability of trial results. Detection of deviations from the predefined quality tolerance limits should trigger an evaluation to determine if action is needed.” Additionally, ICH E6(R2) Section 5.0.7 states that within the clinical study report (CSR), the sponsor should summarize important deviations from the predefined QTLs and remedial actions taken.
Before one can effectively apply QTLs to clinical trials, it is important to understand their unique characteristics relative to historical industry standard terminology such as, for example, clinical trial metrics, key indicators (e.g., key performance indicators, key quality indicators, key risk indicators), parameters, targets, and thresholds.
Metrics are measurements of data that may be applied to numerous categories, including operational timelines, product quality, costs, scientific or medical parameters defined in protocols, and numerous other factors. Key indicators are metrics considered to be important in the conduct and control of a study or program and may relate to performance, quality, or risks. Key indicator thresholds, or targets, are designed to be relatively sensitive in order to detect potential risks or issues that may be mitigated or minimized. If a risk indicator threshold is reached, an appropriate action is taken.
In contrast, QTLs for the purpose of this article will refer to the requirements as outlined by ICH E6(R2) and not any broader definitions. The guideline clearly states QTLs are used “to identify systematic issues that can impact subject safety or reliability of trial results.” A QTL is a level, point, or value associated with a parameter that should trigger an evaluation to determine if there is an issue, especially if subject safety or data integrity are at risk.
It is important to note that a QTL is not a parameter; rather, it is the limit for a parameter whereby safety for subjects becomes impacted beyond appropriate levels, or reliability of trial results is compromised. Therefore, since ICH E6(R2) requires that QTLs be established before a protocol is implemented – so that they may be monitored during the conduct of the trial – it is critical to define the parameters and their units of measure as well as provide justifications for the selection of the parameters and the chosen limits.1 While not all-inclusive of the entirety of parameters that may warrant the establishment of QTLs, categorically-chosen parameters frequently include those related to protocol efficacy and safety measures (primary and possibly secondary endpoints), as well as inclusion/exclusion criteria for patient retention, as these may significantly affect patient safety or reliability of results.
Tolerance limits may be one-sided as depicted within Figure 1,1 or it may be possible to develop two-sided symmetric or two-sided asymmetric tolerances. While less likely, it is also possible to establish a zero-tolerance QTL.
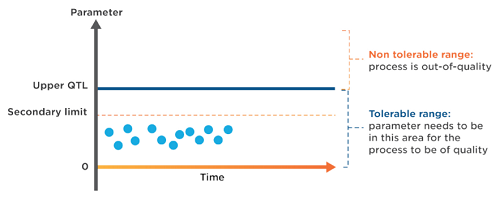
Figure 1: Parameters, QTLs, and secondary limits
Secondary limits may be established as indicators that the parameter metric is approaching the QTL. These secondary limits are intentionally-chosen thresholds whereby a study team may implement strategies and mitigation steps designed to reduce the probability that the QTL will be reached. These secondary limits also provide opportunities to escalate and inform stakeholders that important subject safety or data integrity parameters are at risk.
While these concepts and definitions may be logical and seem somewhat straightforward in theory, many organizations and study teams become quite anxious when implementing QTLs in actual practice. One major reason relates to the implications that result should a tolerance limit be reached during the conduct of a trial. It requires formal actions that may include study suspension or discontinuation, removing patients from treatment, or even determining that study results are not viable. During protocol design, scientific or medical knowledge regarding treatment risks may not be fully known. Therefore, it can be quite difficult to reliably establish limits that represent true subject safety concerns or real impact to interpretability of data.
This prompts the question about appropriate reasons to modify QTLs during the conduct of a trial following study start. Is this acceptable? While not ideal, and preferably should not be done frequently, there are valid reasons for adjusting QTLs and/or the parameters. Limited scientific or medical information about a therapeutic agent in development or incomplete understanding of the mechanism of action may cause incorrect initial assumptions. In some trials, the number of required subjects is uncertain at the beginning of a trial (such as trials to assess the maximum tolerated dose) such that subject retention requirements may be unknown. New scientific information learned while a clinical trial is progressing, either through other ongoing clinical or nonclinical studies by the sponsor, by other companies, or by academia, may warrant a change in the QTL and, in exceptional circumstances, the parameter. In some instances when uncertainty exists while determining QTLs for a parameter, it may be appropriate to engage an independent party (e.g., a Data Safety Monitoring Board (DSMB)) to determine if a valid adjustment would be appropriate.
Valid modifications of any parameter associated with a QTL, or of the QTL itself, during a trial must be justified with documented decisions and properly reported. It is recommended that this be done via formal change control processes and events within an organization’s quality management system (QMS).
In summary:
- QTLs are established during the planning stages of a trial.
- QTLs identify if systematic issues are occurring that may impact subject safety or reliability of trial results.
- When a QTL is exceeded, an evaluation of the deviation is triggered.
- Important deviations and the associated parameters and QTLs are reported in the CSR, along with any remedial actions taken.
- If adjustments to QTLs during the conduct of a trial are considered, it should be recognized that they are the exception and must be justified, documented, and reported.
References:
- Risk-Based Quality Management: Quality Tolerance Limits and Risk Reporting, TransCelerate BioPharma, Inc. http://www.transceleratebiopharmainc.com/wp-content/uploads/2017/09/Risk-Based-Quality-Managment.pdf
About The Author:
 Steve Whittaker is a senior consultant for The Avoca Group. His experience through years of drug development leadership roles, combined with his established professional network across these industries, provides unique and valuable insights for organizational leaders. Whittaker served as executive director of the Avoca Quality Consortium from 2011 to 2018, and for 14 consecutive years on the Advisory Board for the annual Partnerships in Clinical Trials program, chairing the board for two years. In 2009, Steve retired from Eli Lilly and Company, where he served as COO/sr. director of operations and project management for the Cardiovascular/Acute Care Platform. You can connect with him on LinkedIn.
Steve Whittaker is a senior consultant for The Avoca Group. His experience through years of drug development leadership roles, combined with his established professional network across these industries, provides unique and valuable insights for organizational leaders. Whittaker served as executive director of the Avoca Quality Consortium from 2011 to 2018, and for 14 consecutive years on the Advisory Board for the annual Partnerships in Clinical Trials program, chairing the board for two years. In 2009, Steve retired from Eli Lilly and Company, where he served as COO/sr. director of operations and project management for the Cardiovascular/Acute Care Platform. You can connect with him on LinkedIn.