
The Evolving Role Of Biomarkers In Oncology Clinical Trial Design
By David Parker, Ph.D., SVP, diagnostics solutions, Precision for Medicine

In the first installment of this series, we explored payers’ increasing willingness to provide guidance on clinical trial design and pharma’s increasing desire to seek it. With billions of dollars potentially at stake for each party, there is growing awareness that pharma and payers cannot afford missteps in the drug development process. The traditional arm’s-length (or even adversarial) relationship between drug developers and payers is not ideal for both financial success and patient welfare.
The drive for increased value is pushing payers and drug developers toward greater interaction on clinical trial designs. From the payer perspective, this means getting more clinical and cost-of-care benefit for every dollar of drug spend. For the pharma company, it means getting more drug revenue for every dollar invested in drug development. Working more cooperatively and leveraging advances in basic and translational research makes it possible for both stakeholders to achieve their goals.
As the healthcare paradigm shifts from volume to value, payers are seeking greater value from pharmacotherapy by reducing the drug spending required to deliver a given amount of clinical or quality-of-life benefit to a patient. In pursuing this goal, payers have followed two pathways to success.
The first, and more traditional, route is by negotiating better pricing for drugs prescribed to a broad group of clinically eligible patients (or requiring use of low-cost generic equivalents, which has the same net effect). This approach recognizes that some patients may respond weakly to a drug, but attempts to mitigate the wasted value resulting from such weak responses by reducing the drug’s cost for all treatment recipients.
The second route, which has become possible in recent years through advances in the scientific understanding of drug mechanisms of action (MOAs) and patient response mechanisms, is by using biomarker tests for high-priced drugs to identify patients who are likely responders and restricting access for patients who are less likely to respond.
Similarly, drug developers are seeking to extract greater value from their pipeline drug candidates and development processes, both in recognition of their payer customers’ needs and in an attempt to mitigate the high cost of late-stage drug candidate failures.
As we discussed in the previous installment, one way they are accomplishing this is by seeking input from payers and incorporating payer-oriented trial design features and value-oriented endpoints into their clinical development plans. This approach can facilitate the demonstration of payer value, but unfortunately is of little help in lowering the risk of late-stage drug failures. Increasingly, pharma companies are trying to address both issues by incorporating biomarker-based patient stratification strategies into their clinical trials.
Drug developers can realize significant clinical and economic benefits by targeting drug therapies with biomarkers to improve response rates and adherence to therapy, while minimizing complications. This approach can meet payers’ value needs by correlating drug response with a measurable biomarker, so that in the market payers can apply utilization management controls based on biomarker test results. This approach also has the potential to meet pharma’s value needs by focusing clinical trial enrollment on likely responders, thereby increasing a pivotal trial’s chances of success.
Value And Science Meet: Targeted Therapy In Oncology
The development of biomarker-targeted therapies has been a key focus of biomedical research in recent years. In 2016, of the 22 new molecular entities (NMEs) and biologics approved by the FDA, 27 percent (six) were targeted therapeutics.1 This demonstrates a marked increase from 2011's 14 percent1 and from 2005, when only 5 percent of approvals were targeted therapies.2 Although the number of targeted therapies as a proportion of overall NME approvals continues to grow, these have been primarily concentrated in oncology, with limited targeting in other disease areas (Figure 1). (Note: This does not include label expansions for drugs that are already approved.)
This focus in oncology has happened for a number of reasons. First, even with recent treatment advances, the clinical unmet need in oncology remains high, especially considering the relatively poor clinical responses and significant adverse event profiles of untargeted chemotherapeutic regimens.
Second, the scientific understanding of cancer and its genomic etiology is more advanced than of any other noninfectious disease, enabling the rational drug targeting of numerous altered cellular mechanisms resulting from tumor-driving genetic mutations.
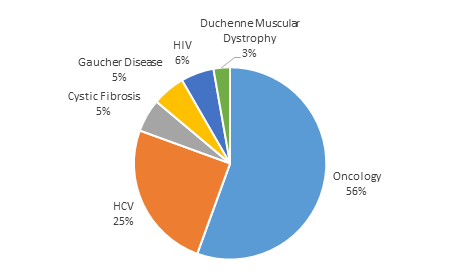
Figure 1: Novel drug approvals with diagnostic-identified target population listed in labeled indication, 2011-2016
Finally, and perhaps most significantly from the perspective of payers and other economic stakeholders, the economic impact of targeted patient selection in oncology is substantial: oncology therapeutics are often very expensive in a concentrated way, creating a highly visible spend where value is at a premium and the economic benefits of targeting therapy to likely-responder patients are obvious.
Therefore, oncology represents perhaps the lowest-hanging fruit for payers to realize value from biomarker-enabled patient stratification for treatment. Because of the high cost of new oncology drugs, these products also present attractive opportunities for biomarkers to improve the value opportunity for their developers: patient targeting can improve the willingness of payers to accept a high per-patient drug cost. In turn, a high per-patient drug cost helps minimize the impact of reducing the size of the treatable population through stratification.
How Can Biomarkers Be Integrated Into Clinical Development To Enhance Value?
In general, a biomarker can be incorporated into a clinical development program in two ways. In the first, genome-wide association studies can be used to identify a biomarker signature that corresponds to robust drug response. Because this approach is indirect (that is, it does not depend on any known biology associating the markers in the signature and the MOA of the drug), it may be affected by multiple factors, including population and geographic variances in biomarker distribution. Therefore, this approach is less likely to deliver value to either the payer or the pharma company.
In the second, more ideal, approach, the biomarker is directly associated with the method of the drug’s action. This is typical for targeted oncology drugs, in which the biomarker may be the drug target itself. Such is the case with epidermal growth factor receptor (EGFR) inhibitors (e.g., erlotinib) and human epidermal growth factor receptor 2 (HER2) inhibitors (e.g., pertuzumab). In other cases, the biomarker may identify another protein in the drug’s action pathway whose alteration enables the tumor to escape the drug’s effect (e.g., RAS mutation as a contraindication for the HER2-targeting drug panitumumab).
In either case, the correlation of the biomarker measurement with drug activity is first determined in nonclinical (in vitro) studies and then undergoes a preliminary assessment in early clinical studies.
Armed with these initial data, the drug developer can design and execute the pivotal trials using a clinical trial assay to include or exclude patients based on a biomarker measurement that indicates likely drug response. This, in turn, establishes the basis for an expectation of enhanced value for the drug developer (higher likelihood of trial success; support for a higher drug price) as well as the payer (biomarker-enabled ability to grant drug access only to likely responders).
Risk Mitigation Collides With Revenue Optimization In Clinical Trial Design
Even when incorporating biomarker stratification into drug development, pharma companies understandably are loath to potentially exclude any patients who might derive clinical benefit from the treatment population. Because individual cancers may have multiple drivers or generate them through tumor evolution, single biomarker targeting is not a perfect predictor of either initial or durable response.
The risk of nonresponse can be reduced by setting a high cutoff for the biomarker, but that comes at the price of excluding some potential responders from the treatment group. This outcome is not desirable for either the drug company, with its interest in optimizing revenue, or the payer, with its interest in seeing the most effective treatment delivered to each patient.
Drug developers have followed two strategies to address this situation. In the first, knowing payers will use the patient’s biomarker test result to determine drug access, drug developers adopt an aggressively low cutoff for the biomarker measurement to exclude as few potential patients as possible. This approach can be successful, particularly when previous studies indicate many patients should have a strong response to drug treatment. However, it is not without risk, as was illustrated by the unexpected failure of Bristol-Myers Squibb’s nivolumab in the CheckMate-026 trial in first-line non-small cell lung cancer treatment. That failure has been attributed to setting the programmed death-ligand 1 (PD-L1) expression cutoff for enrollment too low.
The other strategy some companies adopt involves hedging the bet for success by enrolling a broad population (i.e., without patient stratification), but performing an adequately powered biomarker-based subgroup analysis. This approach can lead to approval based on the subgroup analysis if the primary endpoint is not achieved in the broad population.
Because trials in unstratified populations tend to be larger than those using biomarker-guided enrollment, this approach entails a larger investment in clinical development. However, it enhances the value delivered by increasing the chances of trial success while preserving the possibility of an indication for a broader population.
Clinical Trial Assay To Companion Diagnostic Test
Ultimately, any biomarker test used for patient selection and enrollment in a stratified oncology trial will appear in the drug’s indications for use and therefore must come to market as a commercially available companion diagnostic test. At that point the reimbursement environment for the test itself can strongly influence the success of the drug.
From the pharma perspective, to deliver value a companion diagnostic test must be broadly and easily accessible to all prospectively treatable patients on day one of drug launch. This means the test must be available in a large number of labs and with a relatively short turnaround time, which requires that the labs receive adequate reimbursement for the test and that payers not impose prior authorization or other utilization management controls on the test.
The selection of the biomarker and its assay method in the clinical trial strongly influences whether these goals will be achieved. A biomarker signature that is overly complex or requires measurement with an esoteric assay method may result in a companion diagnostic test that oncologists are reluctant to order, labs are reluctant to offer, and payers are reluctant to reimburse. All of this can negatively influence the drug’s value.
Biomarkers are already a critical element of cancer drug development, and our increasingly deep understanding of the molecular mechanisms of cancer will eventually ensure that all cancer therapeutics will be biomarker-guided. Until then, incorporating biomarkers into oncology clinical trials will be essential to achieving both clinical and commercial goals in today’s value-focused reimbursement environment.
References:
- FDA. New drugs at FDA: CDER’s new molecular entities and new therapeutic biological products. https://www.fda.gov/Drugs/DevelopmentApprovalProcess/DrugInnovation/default.htm. Accessed June 11, 2018.
- Personalized Medicine Coalition. Personalized Medicine at FDA: 2017 Progress Report. http://www.personalizedmedicinecoalition.org/Userfiles/PMC-Corporate/file/PM_at_FDA_2017_Progress_Report.pdf. Accessed June 11, 2018.
About The Author:
 David Parker, Ph.D., is senior VP of diagnostics solutions at Precision for Medicine. He is a strategic visionary and healthcare industry veteran operating at the intersection of reimbursement, health economics, clinical science, and marketing strategy. Parker’s expertise encompasses all aspects of market access, reimbursement, and evidence development strategy, with a particular focus on personalized medicine, advanced molecular and companion diagnostics, and targeted therapies. He has launched successful products, guided major investment and acquisition decisions, and driven favorable coverage and payment determinations by public and private payers alike.
David Parker, Ph.D., is senior VP of diagnostics solutions at Precision for Medicine. He is a strategic visionary and healthcare industry veteran operating at the intersection of reimbursement, health economics, clinical science, and marketing strategy. Parker’s expertise encompasses all aspects of market access, reimbursement, and evidence development strategy, with a particular focus on personalized medicine, advanced molecular and companion diagnostics, and targeted therapies. He has launched successful products, guided major investment and acquisition decisions, and driven favorable coverage and payment determinations by public and private payers alike.