
The "One-Trial" Trap: Why 2026's FDA Efficiency Push Actually Doubles Your Operational Risk
By Johnathon Anderson, Ph.D., associate professor and program officer, UC Davis School of Medicine, and CEO, Peptide Systems

In late 2025, the regulatory ground shifted beneath our feet. FDA Commissioner Dr. Marty Makary announced a fundamental change in the agency’s evidence standards: moving the default expectation for drug approval from two adequate and well-controlled trials to one. With policy updates expected to be finalized within three to six months, this new standard is effectively the reality for any program entering pivotal development in 2026.
On paper, this looks like the efficiency revolution the industry has been clamoring for. The promise of halving your pivotal trial burden suggests faster timelines, reduced recruitment costs, and an accelerated path to commercialization.
But as a clinical operations leader in the cell & gene therapy (CGT) space, where single-trial approvals have some precedent, I have a warning for my peers in broader pharma: Do not confuse efficiency with leniency.
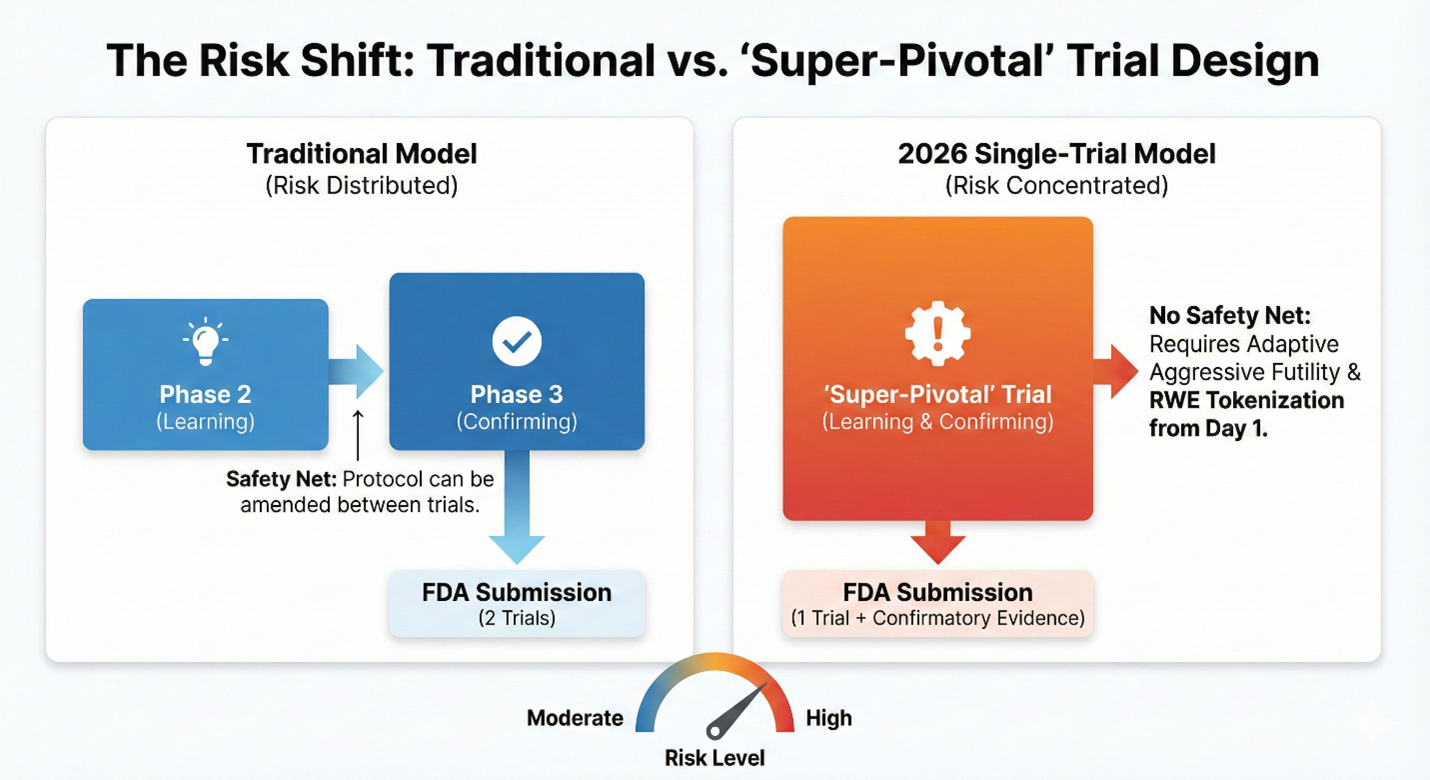
The move to a one-trial standard does not remove the burden of proof; it compresses it. When you remove the safety net of a second confirmatory study, the remaining trial must be operationally flawless. For clinical operations executives, 2026 will not be the year of easier trials. It will be the year of the "super-pivotal" study, a high-stakes operational environment where the margin for error effectively drops to zero.
Here is the strategic road map for navigating this new higher-stakes landscape.
Strategic Pillar 1: Protocol Design For The "Super-Pivotal" Study
The industry often operates under the assumption that a Phase 2 trial is for "learning" and Phase 3 is for "confirming." Under the new single-trial default, this distinction evaporates. Your Phase 2 effectively is your Phase 3.
This creates the "super-pivotal" trial: a study that must be operationally perfect from Day 1.
In the traditional two-trial model, sponsors had a do-over. If the first pivotal trial showed messy data or unexpected variability, you could amend the protocol for the second trial to clean up the signal. That safety net is now gone. In a single-trial submission, a 5% missing data rate isn't just a footnote; it can be the difference between approval and a complete response letter.
The Operational Pivot: Adaptive Efficacy & Broader Confirmatory Evidence
To survive this shift, clinical operations leaders must redesign their protocols to front-load confirmatory evidence. The one-trial pathway is legally predicated on the existence of such evidence, which can range from mechanism-of-action data to animal models.
Operationally, this means the single trial cannot stand alone; it must be scaffolded by a "totality of evidence" package. Sponsors must aggressively integrate these non-clinical and external control data streams (such as natural history studies) into the pivotal submission to satisfy the statutory requirement for "substantial evidence" in the absence of a second trial.
Additionally, sponsors should abandon fixed sample sizes in favor of adaptive design and aggressive futility rules. You should consider writing adaptive boundaries into the protocol that allow sample sizes to increase mid-trial if the effect is lower than modeled. Equally important, you need a mechanism to fail fast and preserve capital if the interim data does not show the definitive signal required for a single-trial approval.
Strategic Pillar 2: Navigating The "Asynchronous" Agency
While the policy landscape is moving faster, the operational machinery of the FDA is facing significant friction. Recent reports of senior leadership departures and workforce reductions signal a profound brain drain at the agency. We are losing the institutional memory that historically facilitated flexible, high-touch negotiation.
We are effectively entering an era of regulatory uncertainty. In previous years, a sponsor could rely on a seasoned division director to steer a program mid-flight. Today, with new leadership and fewer project managers, that lifeline is fraying.
The Internal Risk: Navigating A Divided Agency
Sponsors must also be cognizant of the potential friction between the new deregulatory mandate from leadership and the scientific rank-and-file. Reports suggest that career reviewers were not consulted on some of these swift policy shifts.

The Risk: A reviewer who philosophically disagrees with the single-trial mandate may not openly defy it but could exercise passive resistance through granular technical queries on CMC, biomarkers, or safety signals.
The Strategy: Your data package must do more than just explain your drug; it must arm your reviewer. You need to provide the internal ammunition they will need to justify a controversial single-trial approval to their own skeptics.
The Operational Pivot — Defensive Documentation: The implication is clear: You can no longer plan your timeline around a fix-it meeting with the agency. We are moving from a collaborative dialogue model to an asynchronous review model.
Sponsors must adopt a strategy of defensive regulatory documentation. Because you cannot rely on a project manager to clarify ambiguities via email, your submission packages must be self-explanatory. We are advising teams to include extensive reviewer guides, annotated road maps that link data directly to the endpoint claim, so the reviewer doesn't need to ask questions to find the answer. In 2026, the fastest way through the FDA is not just good science; it is administrative perfection.
Strategic Pillar 3: Real-World Evidence (RWE) Strategy & Budgeting
Perhaps the most significant strategic adjustment for 2026 is understanding that the new efficiency model does not eliminate data requirements; it simply resequences them. Some call this the RWE mortgage.
In exchange for the speed of a single-trial approval, the FDA is levying a heavy tax on the back end: aggressive, long-term post-market surveillance. Recent signals indicate that while the agency is willing to accept less pre-market data, they are countering that flexibility with a demand for "registry-grade" real-world evidence (RWE) to confirm durability over the drug's life cycle.
The Operational Pivot: Tokenization At Source
For clinical operations leaders, this requires a fundamental reallocation of the budget. Resources typically earmarked for a second Phase 3 trial must now transfer to medical affairs for post-approval infrastructure.
To prepare for this "mortgage," sponsors should build their Phase 2 database with tokenization from the very first patient enrolled. This makes your data interoperable by design, allowing you to seamlessly link trial data to real-world claims data post-approval without the costly friction of re-consenting patients years later. By ensuring your clinical trial database can toggle into a real-world registry upon approval, you avoid rebuilding your data engine from scratch.
Precision Over Volume
The FDA of 2026 offers a faster lane to market, but it is a narrower one.
For the industry, the shift from a two-trial to a single-trial standard requires a change in mindset. We must stop viewing Phase 2 as a learning phase and start treating every patient enrolled as a pivotal data point.
In this new era, operational excellence is no longer just about speed or efficiency; it is about precision. The safety net of the second trial is gone. The sponsors who thrive in 2026 will be those who understand that when you only have one shot, you don’t just need good data, you need definitive proof.
Note: The views and opinions expressed in this article are those of the author and do not necessarily reflect the official policy or position of the University of California, Davis, or UC Davis Health.
About The Author:
 Johnathon Anderson, Ph.D., is the associate professor & program officer at the UC Davis School of Medicine. Dr. Anderson brings over a decade of specialized expertise in academic regenerative medicine drug discovery and development, GMP process development, and clinical trial operations. His background includes co-founding startups and securing extensive non-dilutive funding.
Johnathon Anderson, Ph.D., is the associate professor & program officer at the UC Davis School of Medicine. Dr. Anderson brings over a decade of specialized expertise in academic regenerative medicine drug discovery and development, GMP process development, and clinical trial operations. His background includes co-founding startups and securing extensive non-dilutive funding.