
4 Tips For Successful Collaboration With Your EU Qualified Person (QP)
By Enith Morillo and Azeem Shan

Qualified Persons (QPs) are responsible for the certification of clinical trial materials in the EU and therefore are an essential link in U.S. sponsors’ supply chains. Indeed, engaging QPs well before the clinical trial material needs to be certified not only assures a smooth release process and trial start-up but mitigates pitfalls associated with unanticipated technical, quality, and regulatory challenges.
Part 1 of this article covered the QP eligibility process, the difference between the QP Declaration and QP Certification, and QP-to-QP agreements. In this part, more in-depth direction is provided on anticipating QP requirements by involving QPs early on and building a collaborative working relationship that is based on understanding of their role and the legal context they must abide by.
Start With The Drug Substance
Ideally, selection of CMO partnerships for drug substance and drug product manufacture take into consideration where the clinical study may be registered and conducted. However, there are instances in which these business relationships are established long before the study design and country selections are conceptualized.
In Europe, ICH Q7 GMPs for APIs was transferred into the EU regulatory framework in 2010 by amending the EU GMP standards; it can be found in EU GMP Part II, the foundation for drug substances used for clinical trials materials in the EU.
QPs are not strictly required to have physically audited drug substance sites or verify their compliance to EU GMPs as part of the QP Declaration process for Phase 1 clinical trials in the EU, unless the drug substance is sterile. Likewise, QPs are not required to review drug substance executed batch records or release test data for non-sterile drug substances as part of the QP Certification of Investigational Medicinal Products (IMPs). However, this does not exempt U.S. sponsors from due oversight to ensure the drug substance batch and manufacturing site conform to EU GMP standards.
This is an area where U.S. sponsors fall short at times, as the QP will still require written confirmation that each batch of drug substance (used within the manufacture of an IMP): 1) complies with the Investigational Medicinal Product Dossier (IMPD), including BSE/TSE status if not detailed in the IMPD, 2) was manufactured in compliance with EU GMP standards, and 3) was tested to written specification per a certificate of analysis.
In cases where the drug substance is sterile, EU QPs are required to verify that the full non-EU supply chain detailed within the IMPD and, consequently, the QP Declaration complies with EU GMP standards, including drug substance manufacturing and bulk drug product filling sites. In addition, for QP certifications involving sterile drug substances, QPs are required to review GMP documentation, such as executed batch records, certificate of analysis, information on in-process-testing and filter integrity testing, autoclave cycle data, lyophilizer cycle data, etc., along with environmental monitoring data for all stages of the process. Additional documentation may be requested on a case-by-case basis, dependant on the unique characteristics of the molecule (e.g., biologics).
Bypass Repeat Audits
Although QPs are not required to audit non-sterile drug substance manufacturing sites for materials intended for clinical trials, it is recommended that U.S. sponsors engage experienced auditors or QPs to perform such audits as part of their vendor management program. This will ensure the audits appropriately assess how EU GMPs are applied to early stages of development and that technology transfer considerations are included in such assessments in anticipation of scale-up requirements.
As part of the QP Declaration process, QPs are required to verify that non-EU drug product manufacturing sites are compliant with EU GMPs.
QPs may approach the verification process in several ways:
- The QP completing the QP Declaration and performing the QP Certification of the batch of IMP performs an independent audit of the non-EU manufacturing site(s) for compliance with EU GMPs.
- The sponsor provides an audit report of the non-EU manufacturing site to the QP who will be completing the QP Declaration and performing the QP Certification of the batch of IMP.
- The QP completing the QP Declaration and performing the QP Certification of the batch of IMP contractually works with a peer QP who confirms the non-EU manufacturing sites meet EU GMPs, among other requirements.
In general, audits of the non-EU manufacturing sites are performed well before a QP is engaged by the U.S. sponsor. U.S. sponsors must ensure that these audits are conducted in a manner that guarantees acceptance by the certifying QP. Therefore, it is highly advised that U.S. sponsors:
- Engage auditors who are knowledgeable and have experience performing audits against EU GMPs or, alternatively, contract QPs to conduct such audits.
- Request audit reports be well-written, thorough, and cover the full scope of services related to the IMP to be imported into the EU.
- Oversee satisfactory resolution of audit findings that may call into question the compliance of the site or IMP with EU GMPs.
The QP signing the QP Declaration must have confidence on these items to attest to the EU GMP compliance of the site.
By proactively implementing these recommendations, U.S. sponsors maximize the possible acceptance of said audit reports by the QP completing the QP Declaration and avoid the potential delays and added expenses that may be incurred by repeat audits.
Strategize Beyond Checklists
QP checklists are generally provided by CMOs to U.S. sponsors as a running list of the documentation the QP certifying the batch of IMP will need to review. However, these checklists are intended as guidance on the minimum requirements for QP certifications and may not be all-inclusive.
Instead of approaching QPs with a checklist in hand, U.S. sponsors are strongly encouraged to build a collaborative and knowledge-based relationship with the QPs they work with, keeping in mind the shared responsibility for patient safety a QP assumes when certifying a batch of IMP for an EU clinical trial.
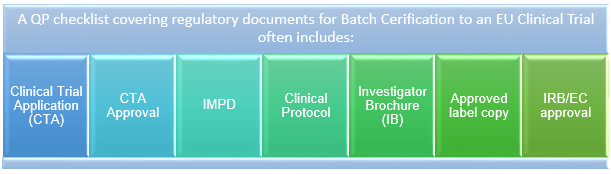
Figure 1: QP checklist covering regulatory documents for batch certification to an EU clinical trial
Significant delays in the QP certification of a batch may at times result from poor communication and misaligned expectations during the onboarding and release process.
For example, although seemingly trivial, the documents listed in Figure 1 are required for each EU Member State participating in the clinical trial and, as multiple languages are involved, verified translations are required.
Likewise, QPs certify batches of IMP against the most current version of the approved CTA, which has been submitted to each competent authority. Thus, it is imperative for sponsors to adequately manage version control of documents throughout the clinical life cycle. QPs may also request to see communications between sponsors and competent authorities that directly impact the CTA submission or amendments in relation to certification of a batch.
Building a working relationship with the QP goes beyond checklists and proactively facilitates a smooth batch certification process.
Avoid Shipping Excursions
IMPs used for Phase 1 studies are commonly supported by limited and ongoing stability studies for both the establishment of an expiration date and assessment of temperature excursions. At times, shelf-life predictions and extrapolations are also incorporated in setting expiry dates, in alignment with ICH Q1 Stability Testing of New Drug Substances and Products.
To support continued supply to patients, expiry date extensions are generally necessary as additional stability data becomes available, requiring QP recertification of the batch(es) in question.
However, when it comes to temperature excursions, QPs do not have the same flexibility as their U.S. QA counterparts to evaluate and interpret available data to determine suitability of IMPs for distribution. Rather, QPs are expected to rely on the sponsor and the stability profile within the IMPD to assess the suitability of the affected IMP.
Thus, U.S. sponsors must oversee the proper compilation of the IMPD and mitigate the risk of excursions through conservative shipping arrangements, such as use of validated shippers and temperature monitoring devices, where available. It is also advised that sponsors generate and maintain fit for use reference tables summarizing the available stability data for easy reference in the event of an excursion and that these tables are updated frequently, as additional data becomes available.
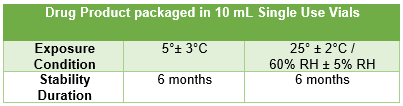
Figure 2: Example of fit for use reference table
Ultimately, it is fundamental U.S. sponsors understand the responsibilities the QP assumes through the QP Declaration and the QP Certification of a batch of IMP to a clinical trial. When sponsors have an appreciation for the shared responsibility for patient safety, and a deeper understanding of the QP’s scope of work, a winning regulatory, quality, and supply chain strategy can be put in place that helps to expedite the clinical timeline and supports uninterrupted clinical supply.
About The Authors:
 Enith Morillo, M.Sc., is the founder and principal consultant of Cadoret Global, a U.S.-based consulting company that specializes in supporting virtual, early stage, and small-sized pharmaceutical companies in taking their investigational drug through development and clinical trials. She has over 12 years of proven, hands-on experience working in the FDA and DEA regulated industry. Morillo specializes in CMC QA oversight of contract manufacturing organizations, batch record review and product disposition, GMP audits, and establishing phase-appropriate quality management systems for emerging pharmaceutical companies. You can reach her at Morillo.Enith@outlook.com or connect with her on LinkedIn.
Enith Morillo, M.Sc., is the founder and principal consultant of Cadoret Global, a U.S.-based consulting company that specializes in supporting virtual, early stage, and small-sized pharmaceutical companies in taking their investigational drug through development and clinical trials. She has over 12 years of proven, hands-on experience working in the FDA and DEA regulated industry. Morillo specializes in CMC QA oversight of contract manufacturing organizations, batch record review and product disposition, GMP audits, and establishing phase-appropriate quality management systems for emerging pharmaceutical companies. You can reach her at Morillo.Enith@outlook.com or connect with her on LinkedIn.
 Azeem Shan, BSc (Hons), MRPharmS, is an experienced EU Qualified Person (QP) based in the UK. A pharmacist by training, he has over a decade of experience in the manufacture, packaging, testing, and distribution of human and veterinary medicines, both as clinical trial materials and commercial products. Shan has first-hand experience with setting up quality management systems, training, performing audits of GDP and GMP facilities, and providing support with remediation issues. His expertise covers EU GMP requirements, including successful MHRA and Veterinary Medicines Directorate inspections, along with site support with FDA pre-approval and GMP inspections. You can reach him at azeem_shan@hotmail.com or connect with him on LinkedIn.
Azeem Shan, BSc (Hons), MRPharmS, is an experienced EU Qualified Person (QP) based in the UK. A pharmacist by training, he has over a decade of experience in the manufacture, packaging, testing, and distribution of human and veterinary medicines, both as clinical trial materials and commercial products. Shan has first-hand experience with setting up quality management systems, training, performing audits of GDP and GMP facilities, and providing support with remediation issues. His expertise covers EU GMP requirements, including successful MHRA and Veterinary Medicines Directorate inspections, along with site support with FDA pre-approval and GMP inspections. You can reach him at azeem_shan@hotmail.com or connect with him on LinkedIn.