
Understanding The Basics Of EU Clinical Trials With CTIS
By Guriqbal Singh and Kamil Shaik, RegWeb Consulting Services Inc.

In the rapidly evolving healthcare sector, where advancements have the potential to revolutionize lives and our comprehension of medicine, the EU has made a significant stride by implementing the Clinical Trial Regulation (EU) 536/2014 (CT Regulation), which replaces the old Clinical Trials Directive 2001/20/EC (CT Directive).
Clinical Trials Information System (CTIS) streamlines the process for evaluating and overseeing clinical trials across the EU. A unified clinical trial application (CTA) with a core dossier is submitted for approval, reaching all European national competent authorities and ethics committees and ensuring the trial is registered in a public database.
Before CTIS, clinical trial sponsors needed to submit separate applications to national competent authorities and ethics committees in each country to obtain trial approval. The new CTIS system tackles issues like the fragmentation and complexity of clinical trials, lack of transparency, administrative burdens, slower approval processes, and the limited harmonization seen under the previous CT Directive.
Key advantages of CTIS include:
- a single integrated submission system that enables European member states to collaborate and coordinate in the evaluation and supervision of clinical trials
- more efficient and transparent regulation of clinical trials
- a user-friendly website that allows the public to search for information on all clinical trials approved through CTIS
- reduced administrative burden for researchers and pharmaceutical companies
- facilitated patient recruitment by making clinical trial information accessible to patients
- faster and more coordinated clinical trial authorization
- high safety standards for all participants in EU clinical trials
- fully electronic exchange of information between sponsors and regulatory agencies
- the possibility to extend clinical trials to additional member states to improve recruitment rates.
Transition Period
Starting on Jan. 31, 2022, the CT Regulation replaced the CT Directive, marking the beginning of a three-year transition period that will conclude on Jan. 30, 2025.

Clinical trials authorized under the CT Directive that are expected to continue in at least one EU/EEA member state beyond Jan. 30, 2025, must be transitioned to CTIS.
Getting CTIS Access
To access the CTIS, sponsors must have an active EMA account. Sponsors already using other EMA applications such as Eudralink, SPOR, IRIS, Eudravigilance, or OMS can access CTIS with their existing accounts. New users can create an EMA account through self-registration.
Submitting A CTA
For a clinical trial, three types of applications can be submitted through CTIS:
- Initial CTA: A request to conduct a clinical trial that includes comprehensive information about the trial for evaluation by the Member State Concerned (MSC).
- Additional MSC CTA: A request by the sponsor to extend an authorized clinical trial to one or more additional MSCs.
- Substantial Modification CTA: A request by the sponsor to make changes to the clinical trial that could significantly impact the safety or rights of the participants or the reliability of the generated data.
Sponsors can also submit non-substantial modifications during an ongoing clinical trial. These modifications are not considered applications, as they are not evaluated by the MSCs.
Submitting an Initial CTA
Creating a new CTA involves completing the following steps:
- Forms: This section includes administrative information such as a cover letter and proof of payment.
- MSC: This section includes information related to the MSCs and the number of subjects targeted for each member state.
- Part 1: This section includes details concerning the trial, sponsor, product, and all trial-specific documents.
- Part 2: This section includes country-specific details such as trial sites, facilities, and recruitment details in each MSC.

The sponsor must complete these sections to file an initial clinical trial application.
Overview Of The Process And Timelines For An Initial CTA
For clinical trials conducted in multiple EU countries, the sponsor proposes a Reporting Member State (RMS) from among the MSCs when submitting the initial application dossier. The final RMS is assigned after the RMS selection tasks are completed by all MSCs.
The MSC proposed by the sponsor to be the RMS will indicate its willingness or unwillingness to take on this role. If the proposed MSC is unwilling, an RMS will be chosen from the pool of MSCs that are willing to serve in this capacity.
The selected RMS will play a leading role in the evaluation process. The RMS leads the evaluation, consolidates considerations during the validation and Part I assessment phases, and issues the conclusion on Part I.
In a single-nation trial, the RMS is assigned to the sole MSC selected by the sponsor in the application.
According to the CTR, the entire initial CTA evaluation by the MSC should be completed within 60 days. However, this timeline may be extended if the MSC raises a Request for Information (RFI) during the evaluation process.
The 60-day timeline can be extended by up to 15 days for Validation RFIs and up to 31 days for Assessment RFIs.
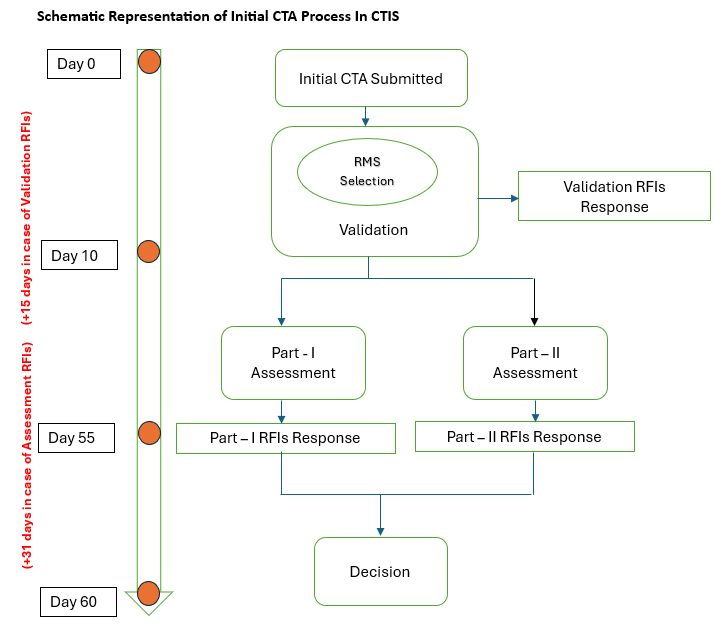
Note: CTIS incorporates a dynamic workflow that recalculates the deadlines for subsequent tasks/actions if a task/action is completed ahead of its deadline. For certain tasks performed by MSCs, earlier completion can shorten the timelines for the following tasks/actions, potentially resulting in a shorter overall evaluation period.
Sponsors who fail to respond to RFIs raised by MSCs before the given deadline in the system will cause their application to lapse.
References:
- EMA Clinical trials’ transition to new EU system, clinical trials website - EMA. Available at: https://euclinicaltrials.eu/about-this-website/#transition-period.
- European Medicines Agency Clinical trials information system, Clinical Trials Information System | European Medicines Agency. Available at: https://www.ema.europa.eu/en/human-regulatory-overview/research-and-development/clinical-trials-human-medicines/clinical-trials-information-system
- Ctis evaluation timelines - european medicines agency (2024) CTIS Training Program. Available at: https://www.ema.europa.eu/en/documents/other/clinical-trial-information-system-ctis-evaluation-timelines_en.pdf.
- Quick guide - transitional trials from EudraCT to CTIS (2024) CTIS Training Programme. Available at: https://www.ema.europa.eu/en/documents/other/sponsors-guide-transition-trials-eudract-ctis-ctis-training-programme-module-23_en.pdf.
- Step-by-step guide (2021) how to evaluate an initial CTA - RMS Selection. Available at: https://www.ema.europa.eu/system/files/documents/other/cttm06_step-step_guide_en.pdf
About The Authors:
 Guriqbal Singh is the founder and director of RegWeb Consulting Services Inc., based in Canada. Regweb provides solutions for complex submission strategies concerning CMC and clinical research aspects. Regweb provides end-to-end services for pre-authorization to post-authorization stages of a molecule's life cycle. Regweb is also involved in advertisement and promotional material review along with systematic literature search based on the Cochrane method.
Guriqbal Singh is the founder and director of RegWeb Consulting Services Inc., based in Canada. Regweb provides solutions for complex submission strategies concerning CMC and clinical research aspects. Regweb provides end-to-end services for pre-authorization to post-authorization stages of a molecule's life cycle. Regweb is also involved in advertisement and promotional material review along with systematic literature search based on the Cochrane method.
 Kamil Shaik is a regulatory affairs specialist at RegWeb Consulting Services Inc. He holds a master's degree in regulatory affairs and has experience in the pharmaceutical industry. His expertise spans CMC, regulatory intelligence, and project management.
Kamil Shaik is a regulatory affairs specialist at RegWeb Consulting Services Inc. He holds a master's degree in regulatory affairs and has experience in the pharmaceutical industry. His expertise spans CMC, regulatory intelligence, and project management.