What Is Computer Software Assurance, And What Are Its Benefits?
By Kamila A. Novak, KAN Consulting

Ever since the FDA published guidance on computer system validation (CSV) in 20221, it’s become a daunting task for regulated industry, which has often chosen to screenshot each step of every test case —no matter how trivial — as a standard approach to documenting validation activities. Although the General Principles of Software Validation already included a risk-based approach to validation, most organizations did not implement it, likely due to uncertainty about necessary documentation and compliance concerns. Thus, the laborious documentation-heavy approach has persisted for the past two decades.
A breakthrough occurred in September 2022, when the FDA released the draft guidance Computer Software Assurance (CSA) for Production and Quality System Software and finalized its latest update in February 2026.2 The FDA’s goal is to help manufacturers produce high-quality medical devices while complying with the QMSR (21 CFR Part 820).3
Since then, ISO standard 13485:2016 has been the basis of 21 CFR Part 820, which requires medical device manufacturers to establish quality systems for the design, manufacture, packaging, labeling, storage, installation, and servicing of finished devices to ensure safe, effective, and compliant products. ISO 13485 and the QMSR are specific for medical devices and do not apply to other regulated areas that typically have their frameworks, such as current cGMPs, ICH Q10 Pharmaceutical Quality System, etc.
What Is Computer Software Assurance (CSA)?
CSA is a risk-based approach for establishing and maintaining confidence that software is fit for its intended use. The guidance provides a risk-based framework for software assurance, examples of various testing methods, and many use cases on how to apply it. In addition, the CSA guidance recommends leveraging validation performed by software vendors and outlines additional assurance considerations for organizations using such software.
The latest version now includes the following sections:
- Definitions of cloud computing, Infrastructure as a Service (IaaS), Platform as a Service (PaaS), and Software as a Service (SaaS)
- Production or Quality Management System Software Changes
- Additional Considerations for Assurance Activities
- Considerations for Electronic Records Requirements
- Example 4 SaaS Product Life Cycle Management System
CSA Guidance Deep Dive
Let us look closer at each section of the CSA guidance and summarize its key points.
Section V. A. CSA Risk Framework
The CSA guidance describes a risk-based approach to establish confidence in the automation used for production or quality management systems, to identify where additional rigor may be appropriate, and various methods and testing activities that may be applied to establish computer software assurance. The FDA’s goal is to help manufacturers produce high-quality medical devices while complying with the QMSR (21 CFR Part 820).
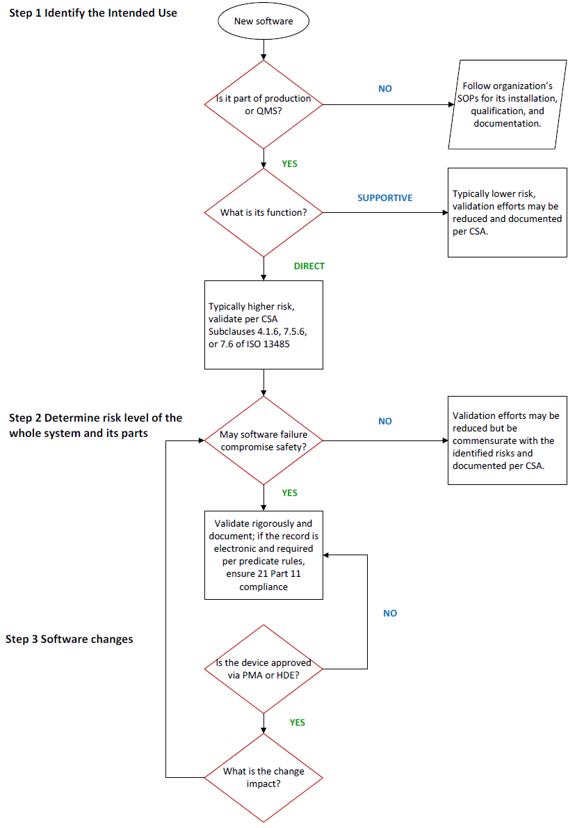
The CSA guidance uses a simple binary risk categorization, high risk versus non-high risk, although the FDA acknowledges that manufacturers may follow a more granular approach to risk categories, e.g., medium, low, etc. The risk framework follows six main steps:
- Identify the system’s intended use.
- Determine the risk-based approach based on factors with potential impact on the computerized system performing as intended.
- Manage production or quality management system software changes.
- Determine appropriate assurance activities commensurate with risk, e.g., types of testing depending on the risk level.
- Consider additional assurance activities for systems supplied by vendors.
- Establish adequate records, including sufficient objective evidence to demonstrate the software performs as intended.
To determine the level of assurance effort and activities appropriate to establish confidence in the software, risk assessment should focus on potential compromised safety and/or quality of the device. The CSA follows the least burdensome approach, where the burden of validation is no more than necessary to address the risk.
Section V. A. (1) Identify System’s Intended Use
The first step is to determine if the software will be used directly for manufacturing or quality management, or if its function will be supportive. Supportive software usually presents lower risk; hence, validation efforts may be reduced.
Organizations utilize not only software specific to production or the quality management system, but also software intended for management of general business processes or operations, such as email or accounting applications, and software intended for establishing or supporting infrastructure, such as networking, user authentication, or continuity of operations (e.g., backup and restore). While the CSA guidance does not apply to these systems, their risks related to business criticality, cybersecurity, confidentiality, etc., should be assessed and categorized (e.g., using GAMP 5)4, and the software should be adequately validated to protect the organization’s business.
The decision process of determining the intended use should be documented since the use and deployment, especially for cloud computing systems or commercial-off-the shelf (COTS) software, may have various use cases.
Section V. A. (2) Determine The Risk-Based Approach
This risk-based approach includes the systematic identification of reasonably foreseeable software failures, which determines whether such a failure poses a high process risk, and selecting and performing assurance activities commensurate with the medical device or process risk. The risk-based analysis for production or quality management system software focuses on factors that may impact or prevent the software from performing as intended, such as proper system configuration and management, system security, data integrity, data storage, data transfer, or operation error. This is different from performing a risk analysis for a medical device as described in ISO 14971:2019 – Medical devices – Application of risk management to medical devices.5
The CSA guidance discusses both process risks and medical device risks. A process risk refers to the potential to compromise production or the quality management system, while a medical device risk refers to the potential for a device to harm the patient or user. Process risks should establish whether the process itself is ahigh risk, such as maintaining process parameters like temperature or humidity, or non-high risk, e.g., corrective and preventive actions (CAPA) routing, automated logging/tracking of complaints, etc.
Section V. A. (3) Manage Software Changes
This section provides reporting expectations for changes of software used in production or quality management and applies to devices with approved premarket approval applications (PMA) or humanitarian device exemptions (HDE).
The guidance includes an example of a manufacturing execution system (MES) within medical devices and within the scope of PMA/HDE contexts. If it is used to manage workflow, track progress, record data, and establish alerts or thresholds based on validated parameters that are part of maintaining the quality management system, a failure to perform as intended may disrupt operations but not affect the process parameters established to produce a safe and effective device. Generally, changes affecting these MES operations are to be submitted in annual reports. In contrast, changes in MES used to automatically control and adjust established critical production parameters, such as temperature, pressure, or process time, may change a manufacturing procedure that affects the safety or effectiveness of the device. In this case, changes are to be submitted via 30-day notice.6
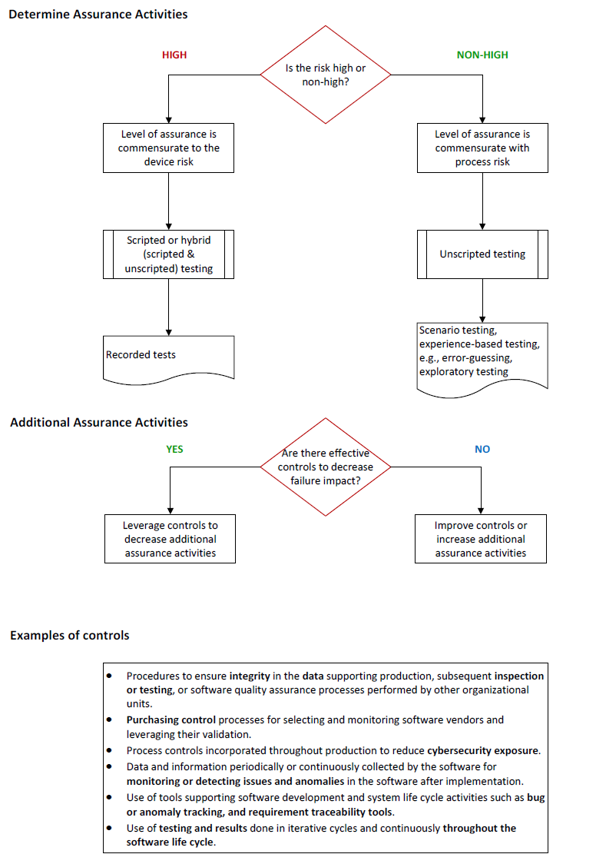
Section V. A. (4) Determine Appropriate Assurance Activities
Once the organization has established whether a software feature, function, or operation poses a high process risk, i.e., a quality problem that may foreseeably compromise safety, it should determine assurance activities commensurate with the medical device risk or the process risk:
- If the quality problem may foreseeably compromise safety (high process risk), the level of assurance rigor should be commensurate with the medical device risk.
- If the quality problem may not foreseeably compromise safety (non-high process risk), the level of assurance activities should be commensurate with the process risk.
In both cases, increased risks generally require greater rigor for assurance, i.e., a greater amount of objective evidence, and relatively low risk (non-high process risk) generally implies a lower amount of objective evidence for assurance activities.

The CSA guidance describes manual and automated testing options including scripted testing (test cases are recorded) and unscripted testing (dynamic testing in which the tester’s actions are not prescribed by written instructions in a test case). The latter can be executed based on scenarios (ad hoc testing) or experience (error guessing and exploratory testing).
Unscripted testing does not mean testing without any documentation. However, unlike traditional screenshots used to document validation activities, unscripted testing may include specification-based test case design techniques based on exercising sequences of interactions between the test item and other systems and testing concepts such as test attacks, tours, and error taxonomies that target potential problems such as security, performance, and other quality areas.
Section V. A. (5) Additional Assurance Activities
This section outlines additional controls that may decrease the impact of compromised safety and quality if failure of the software feature, function, or operation were to occur. Such controls include but are not limited to:
- procedures to ensure integrity in the data supporting production, subsequent inspection or testing, or software quality assurance processes performed by other organizational units
- purchasing control processes for selecting and monitoring software vendors and leveraging their validation
- process controls incorporated throughout production to reduce cybersecurity exposure
- data and information periodically or continuously collected by the software for monitoring or detecting issues and anomalies in the software after implementation
- use of tools supporting software development and system life cycle activities, such as bug or anomaly tracking, and requirement traceability tools
- use of testing and results done in iterative cycles and continuously throughout the life cycle of the software.
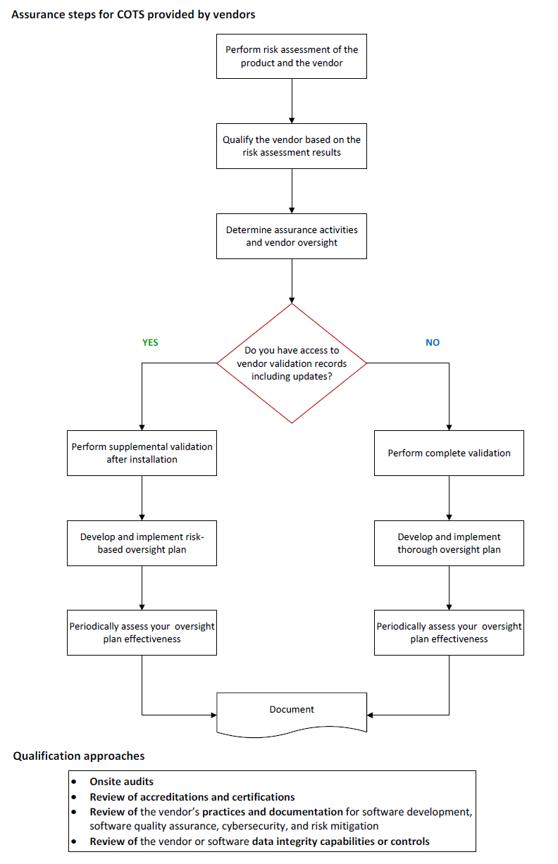
This section provides guidance and recommendations for systems and software supplied by vendors. Organizations should focus on rigorous vendor selection and (re)qualification, including audits of vendors providing high-risk systems. Also, organizations can leverage validation and assurance performed by vendors supposing vendors provide adequate evidence. With appropriate justification and risk assessment, organizations can reduce their assurance activities.
Section V. A. (6) Establish Adequate Records
Organizations should capture sufficient objective evidence to demonstrate that the software feature, function, or operation was assessed and performs as intended. Records should include:
- the intended use of the software feature, function, or operation
- the result of the risk-based analysis of the software feature, function, or operation
- documentation of the assurance activities conducted:
- A description of the testing conducted based on the assurance activity
- Issues found during testing, e.g., deviations, defects, and/or failures
- A conclusion statement declaring acceptability of the software for its intended use. If issues were found, resolution of issues should be part of the conclusion statement, e.g., process controls implemented to address any impact from the issues to the intended use or appropriate risk justification addressing why the issues found will not impact the intended use.
- Record of who performed testing/assessment and date it was performed
- Established review and approval when appropriate, e.g., a signature and date of an individual with signatory authority
The record should retain sufficient details of the assurance activity to serve as a baseline for improvements or as a reference point if issues occur.
Section V. B. Considerations For Electronic Records Requirements
Any electronic records and signatures for regulatory purposes should comply with 21 CFR Part 117 or Annex 11, currently being revised, in the European Medicines Agency jurisdiction.8
For computer software used as part of production or the quality management system, the applicable predicate rules include those under Part 820. A document required under Part 820 (including but not limited to a document required to be signed) and maintained electronically would generally be an “electronic record” under Part 11 (see 21 CFR 11.3(b)(6)). To determine when a record is required under Part 820, organizations should consider if the record is necessary as evidence of validation. If an organization maintains a document required under Part 820 in electronic form, then Part 11 generally applies.
Key Benefits Of CSA
Transitioning from traditional CSV to CSA offers organizations several benefits:
- Reduced documentation burden: Instead of the traditional one-size-fits-all approach to validation documentation, CSA encourages organizations to focus on areas where software failure may jeopardize patient safety and/or product quality.
- Savings: By leveraging unscripted testing, ad hoc methods, and vendor-provided testing results, organizations can significantly save resources spent on validation.
- Agility and scalability: The risk-based model integrates seamlessly with modern development methodologies like Agile and DevOps. Organizations can deploy cloud-based tools and new software updates faster.
- Improved patient safety and quality: By reducing paperwork, quality and compliance teams can focus on areas that genuinely protect product quality and consumer safety.
- Streamlined inspection readiness: Documenting risk-based decisions and their rationale and utilizing real-time automated traceability create a more transparent validation process.
- Stronger regulatory alignment: Following CSA principles and recommendations helps establish objective evidence that software is fit for its intended use, reducing potential compliance challenges.
See the decision trees in Appendix 1 that organizations can follow when implementing CSA in validation processes.
Conclusion
The final CSA guidance document outlines practical steps and examples for implementing a risk-based approach to software validation that aligns with the FDA guideline General Principles of Software Validation, the FDA Part 11 Scope and Application Guidance Document, and with the broader regulatory shift toward ISO 13485 harmonization under the amendments to 21 CFR 820 QMSR.1,9,3 It provides recommendations for fit-for-purpose assurance testing and documentation that help organizations utilize their resources better while ensuring that systems and software they deploy are and remain fit for their intended use.
References:
- General Principles of Software Validation, FDA, January 2002, https://www.fda.gov/regulatory-information/search-fda-guidance-documents/general-principles-software-validation
- Computer Software Assurance for Production and Quality Management System Software, Final Guidance, February 3, 2026, https://www.fda.gov/media/188844/download
- 21 CFR Part 820 Quality Management System Regulation, FDA, February 2026, https://www.federalregister.gov/documents/2024/02/02/2024-01709/medical-devices-quality-system-regulation-amendments
- GAMP® 5: A Risk-Based Approach to Compliant GxP Computerized Systems, ISPE, 2022 (second edition)
- ISO 14971:2019 – Medical devices – Application of risk management to medical devices
- 21 CFR 814.39(b), 814.108, and 814.126(b)(1), and the “Annual Reports for Approved Premarket Approval Applications (PMA)” guidance
- 21 CFR Part 11 Electronic Records; Electronic Signatures, FDA, https://www.ecfr.gov/current/title-21/chapter-I/subchapter-A/part-11
- EudraLex, The Rules Governing Medicinal Products in the European Union, Volume 4, Good Manufacturing Practice, Medicinal Products for Human and Veterinary Use, Annex 11: Computerised Systems https://health.ec.europa.eu/document/download/8d305550-dd22-4dad-8463-2ddb4a1345f1_en?filename=annex11_01-2011_en.pdf
- Part 11, Electronic Records; Electronic Signatures - Scope and Application, FDA, 2003, https://www.fda.gov/regulatory-information/search-fda-guidance-documents/part-11-electronic-records-electronic-signatures-scope-and-application
About The Author:
 Kamila Novak, MSc, has been involved in clinical research since 1995, having worked in various positions in pharma and CROs. Since 2010, she has run her consulting company, focusing mostly on GXP auditing. She has firsthand experience with countries in Europe, the Middle East, Africa, and North America. Kamila chairs the DIA Clinical Research, Compliance & Quality Community and the SQA Data Integrity Subcommittee, leads the DIA Working Group on System Validation, and serves as a mentor at the SQA and the DIA. In addition, Kamila is a member of the CDISC, the European Medical Writers’ Association, the Florence Healthcare Site Enablement League, the Continuing Professional Development UK, and other professional organizations. She publishes articles and speaks at webinars and conferences. She received the SQA Distinguished Speaker Award in 2023 – 2025, the SQA Distinguished Mentor Award in 2025, and the DIA Global Inspire Award for Community Engagement in 2024. She and her company actively support capacity-building programs in Africa.
Kamila Novak, MSc, has been involved in clinical research since 1995, having worked in various positions in pharma and CROs. Since 2010, she has run her consulting company, focusing mostly on GXP auditing. She has firsthand experience with countries in Europe, the Middle East, Africa, and North America. Kamila chairs the DIA Clinical Research, Compliance & Quality Community and the SQA Data Integrity Subcommittee, leads the DIA Working Group on System Validation, and serves as a mentor at the SQA and the DIA. In addition, Kamila is a member of the CDISC, the European Medical Writers’ Association, the Florence Healthcare Site Enablement League, the Continuing Professional Development UK, and other professional organizations. She publishes articles and speaks at webinars and conferences. She received the SQA Distinguished Speaker Award in 2023 – 2025, the SQA Distinguished Mentor Award in 2025, and the DIA Global Inspire Award for Community Engagement in 2024. She and her company actively support capacity-building programs in Africa.
Appendix 1 Decision Trees