
When Sponsors Can't Explain How Study Data Flows, Inspection Readiness Breaks Down
By Donna Dorozinsky, founder and CEO, Just in Time GCP

Over the past several years, the nature of GCP inspections has shifted in ways that are subtle but consequential for trial sponsors. Regulatory investigators are spending less time reviewing individual documents in isolation and more time evaluating whether sponsors understand how their studies actually operate in practice.
One request now surfaces with increasing frequency during inspections: “Show us how the data flows for this study.”
This is not a request for technical architecture or system diagrams. Regulatory investigators are not asking for detailed data lineage or validation schematics. Instead, they are seeking to understand how protocol requirements were carried out across sites and vendors, how data moved between systems, where accountability changed hands, and how oversight was applied as that data progressed through the study life cycle.
Inspection data help explain why this request matters. FDA inspection activity has increased steadily since 2021, with inspections becoming more consistent, more systems focused, and more reliant on records-based review. Regulatory investigators are no longer searching for isolated errors; they are assessing whether a sponsor can clearly explain how its clinical, quality, and data processes work together to support trial conduct.
What Inspections Are Revealing About Data Flow
Inspection observations increasingly reflect recurring patterns rather than one-off mistakes. Across FDA Form 483 data and inspection experience shared through industry forums, similar gaps appear repeatedly, even among sponsors with established quality systems.
Inspection findings frequently reflect situations where:
- records exist but do not clearly explain what happened in practice
- oversight activities were performed but are difficult to demonstrate over time
- data are distributed across multiple systems, complicating reconstruction of trial execution.
These patterns do not typically point to a lack of effort. Instead, they reflect growing trial complexity, increased reliance on external partners, and fragmented visibility across systems and functions.
Recent regulatory clarification under ICH E6(R3) reinforces this expectation: Sponsors are accountable not only for the systems used in a trial but for demonstrating how data integrity, oversight, and documentation operate together. The ability to explain how study data flows has become a central component of inspection readiness.
When Vendor Oversight Fails, Data Integrity Is At Risk
Recent enforcement actions further underscore why data flow visibility and vendor oversight cannot remain abstract concepts. In 2024, the FDA issued a warning letter to Applied Therapeutics, citing significant concerns related to the handling and destruction of electronic clinical trial data by a third-party vendor. According to the agency, data associated with a clinical study were permanently deleted before the sponsor had secured appropriate archival copies or ensured adequate controls over vendor data retention practices. The sponsor could not fully reconstruct the affected data sets, raising questions about data integrity, oversight, and compliance with regulatory record retention requirements.
This case illustrates a critical point: When sponsors cannot clearly articulate how data moves between systems, who controls it at each stage, and how it is protected throughout the study life cycle, inspection risk escalates quickly. Data flow is not simply an operational diagram; it is a governance framework. Without clear mapping of ownership, retention responsibilities, system interfaces, and oversight checkpoints, sponsors may not identify vulnerabilities until they become regulatory findings.
The Applied Therapeutics action reflects a broader regulatory expectation reinforced under ICH E6(R3): Sponsors remain ultimately accountable for the quality and integrity of trial data, regardless of vendor involvement. Effective oversight requires more than contractual delegation. It requires documented understanding of where study data resides, how it transfers, and how it is safeguarded over time.
What Sponsors Say When Asked To Explain Data Flow
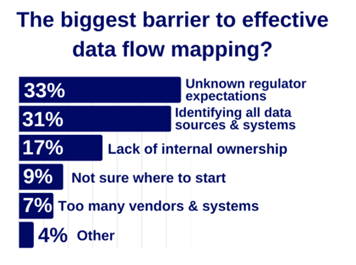
Industry discussions suggest that many sponsors recognize this shift but feel underprepared to respond. In a recent webinar dedicated to this topic, more than 40% of the 140 trialists representing 70 different sponsor companies reported they have been asked by regulatory investigators to explain how their study data flows during an inspection.

Despite understanding this reality, sponsor-side confidence remains limited. Fewer than 10% of respondents indicated they felt confident showing and explaining their study’s data flow under inspection conditions. Many acknowledged that while they expected such questions, they had never aligned internally on how data moved end to end across systems for a single study.
When sponsors described the misalignment and the barriers they encounter, the challenges were rarely technical. Instead, they centered on:
- uncertainty around regulatory expectations
- difficulty identifying all data sources and systems involved in a study
- lack of clear internal ownership across handoffs and vendors.

These themes mirror the same visibility, accountability, and governance gaps that frequently surface during inspections.
How Study Data Flow Maps Uncover Broader Risk
When developed as part of routine trial execution, clinical study data flow maps do more than support inspection responses. They often surface broader quality and operational risks that might otherwise remain hidden.
As sponsors examine how data moves through a study, they frequently uncover:
- unclear ownership at system or vendor handoffs
- data transformations occurring outside validated environments
- inconsistent oversight expectations across partners
- documentation gaps that undermine the ability to explain decisions.
These findings align closely with common inspection observations, including fragmented records, incomplete oversight narratives, and difficulty demonstrating how issues were identified, escalated, and addressed over time.
In this way, study-level data flow mapping functions as a diagnostic and quality-enabling asset. It provides visibility into how trial execution actually occurs, helping sponsors assess whether oversight is applied where risk exists and whether documentation supports the story of the study. Importantly, it also allows teams to identify risks earlier, when corrective action can meaningfully improve trial quality rather than simply respond to inspection findings.
Inspection Readiness Leads To Trial Quality
Inspection readiness in 2026 cannot be achieved through last-minute preparation or document assembly. Rather, sponsors must prepare to clearly explain how trials are conducted and controlled on an ongoing basis. Regulatory investigators are assessing understanding, consistency, and explainability, not documentation volume.
Clinical study data flow maps support this shift by making data pathways, ownership, and oversight touchpoints visible in a study-specific and explainable way. Sponsors who can clearly articulate how their data flows, and why oversight is applied where it is, are better positioned not only to support inspections but to sustain quality across the life of a trial.
As inspections become more frequent, more global, and more records-driven, clarity has emerged as a defining quality asset. The ability to explain how a study operates through its data is central to both inspection readiness and effective trial execution.
References:
- U.S. Food and Drug Administration (FDA). Inspection Observations and Inspection Classification Database. FDA Office of Regulatory Affairs.
https://www.fda.gov/inspections-compliance-enforcement-and-criminal-investigations/inspection-references/inspection-observations
https://datadashboard.fda.gov/oii/cd/inspections.htm - Just in Time GCP. Clinical Study Data Flow Maps Webinar: Survey Results and Industry Discussion. December 2025.
https://hubs.li/Q045fFdT0 - U.S. Food and Drug Administration. Warning Letter to Applied Therapeutics, Inc. (2024). https://www.fda.gov/inspections-compliance-enforcement-and-criminal-investigations/warning-letters/applied-therapeutics-inc-696833-12032024
- European Medicines Agency (EMA). Annual Report on Inspections and Compliance, 2024. https://www.ema.europa.eu/en/annual-report/2024/inspections-and-compliance
- Medicines and Healthcare products Regulatory Agency (MHRA).
UK-Specific Annotations to ICH E6(R3). January 2026. https://www.gov.uk/government/publications/international-council-for-harmonisation-ich-e6r3-annotations
About The Author:
 Founder & CEO of Just in Time GCP, Donna Dorozinsky is a leader and entrepreneur with over 30 years of experience in drug development and clinical compliance. She has expertise in Trial Master File management, clinical operations, safety, data management, regulatory inspection strategy, and more. She is deeply passionate about the business and thinks strategically while paying attention to operational details. Her experience spans small to large pharma and biotech companies, academic centers, CROs, and investigator sites, making her a valuable resource for research organizations seeking clinical compliance assistance.
Founder & CEO of Just in Time GCP, Donna Dorozinsky is a leader and entrepreneur with over 30 years of experience in drug development and clinical compliance. She has expertise in Trial Master File management, clinical operations, safety, data management, regulatory inspection strategy, and more. She is deeply passionate about the business and thinks strategically while paying attention to operational details. Her experience spans small to large pharma and biotech companies, academic centers, CROs, and investigator sites, making her a valuable resource for research organizations seeking clinical compliance assistance.