
ICH E6 R2 – Best Practices For Implementing A More Formal Risk Management Process
By Dr. Volker Hack, Executive Director, Process & System Optimization, and Brian Barnes, Director, Clinical Management, PPD
ICH E6 R2, which was published in November 2016, requires clinical trial teams to implement a more robust risk management process before starting clinical trial activities. The idea is to undertake a well-balanced approach between applying critical thinking and implementing lessons learned. The authors provide several best practices to facilitate implementation of the revised guideline. Besides general guidance to start early, work truly cross-organizationally and have the end in mind, the authors especially highlight the need for a “risk manager,” careful assessment of hybrid scenarios in which tools and processes both from the sponsor and CRO will be used, and the requirement to integrate multiple data sources when rereviewing risks in a clinical trial. While there may be some change management challenges to overcome and extra time and resources to be assigned, the benefits of a robust risk management process designed to improve subject safety and data integrity clearly outweigh any perceived additional efforts.
Introduction
ICH E6 R2, which was published in November 20161, brought a significant shift to clinical trial design and execution because it now requires sponsors and contract research organizations (CROs) to adopt a more formal risk-based approach (RBx) and emphasizes the need for a very robust risk management process2. The quality management section of the revised guideline highlights the principles of risk-based thinking and breaks the concept into two main aspects:
- Defining what is critical to success: During protocol development, identify data and processes critical to human subject protection, as well as the reliability of clinical trial results (“Focus on what matters.”).
- Managing the critical elements of a clinical trial: During the whole life cycle of the clinical trial identify, evaluate, control, communicate, review and report risks.
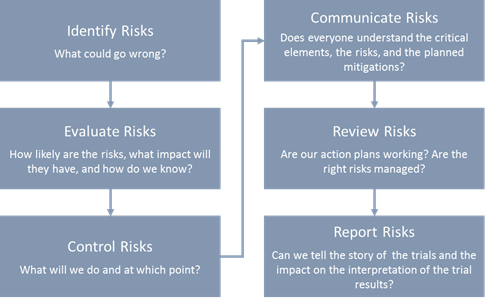
Figure 1: Managing critical elements of a clinical trial
This article focuses on sharing some best practices to overcome potential challenges when implementing the more formal risk management process at a CRO.
Practical Tips
Clinical Trial Setup
Provide adequate support to clinical trial teams: The support of a “risk manager” — an individual whose core competency is risk assessment — has shown to be instrumental especially in the early stages after trial award to drive the risk management process and adequate documentation, ensure quality risk discussions by stimulating critical thinking, review clinical trial risk registers, and facilitate the right decision-making on how risks get mitigated. Risk managers may be established as a new role in a CRO, or responsibilities may be given to a current role (e.g., project manager). If the role of a risk manager is not available, risk management “ambassadors,” who promote risk management principles and support their peers within each of the departments, would be a potential alternative.
Start early: The identification of critical data and processes, as well as any risks to subject safety and data integrity, should start as early as possible — usually when a draft stable protocol synopsis is available — by conducting a thorough and effective risk assessment that enables the clinical trial team to de-risk the protocol (e.g., reduce protocol complexity, reduce ambiguity of inclusion/exclusion criteria, etc.). This early risk management activity also will: support prioritizing resources where it really matters, focus them on the critical elements of the clinical trial from the onset, and ensure that any risk mitigation actions are included in the clinical trial protocol and/or functional plans (e.g., monitoring plan) before they are finalized. Especially when working within a CRO, it is important to understand any risks that were identified upstream (i.e., before the CRO came on board), such as during the creation of the sponsor’s drug development plan.
Balance critical thinking and lessons learned: Several industry consortia have published great tools that help trial teams identify risks (e.g., risk assessment and categorization tool [RACT]). However, these predefined questionnaires and any existing risk libraries (i.e., risks that have been identified by previous trial teams on similar studies) might compromise critical thinking, and clinical trial teams may rely too heavily on these questionnaires and libraries to come up with the critical risks pertaining to their clinical trial. Hence, as discussed earlier, the support of a risk manager, who ensures a well-balanced approach between learning from history and applying risk-based thinking, is of utmost importance to avoid overlooking any unique or previously unidentified risks and ultimately to increase the quality of the risk registers.
Promote cross-organizational risk identification: ICH E6 R2 clearly highlights the need for truly cross-functional risk assessments, which should not only include the different departments within the CRO — best practice is to also involve the sponsor and other vendors, especially if they are collecting data that feed the primary endpoint of the clinical trial (e.g., eCOA vendors) or have an impact on subject safety.
Set appropriate thresholds: ICH E6 R2 also introduced the concept of predefined quality tolerance limits (QTLs) to help identify systematic, protocol-level issues that can impact subject safety or reliability of trial results. It is best practice to establish only a few QTLs (3 to 5) as part of the risk management activities. While setting up QTLs, the study design and the medical and statistical characteristics of the variables being targeted for QTL monitoring are important to consider, since QTLs are intended to target bias in trial conduct or systemic errors that can be traced back to a single root cause. The targets for QTL monitoring should correspond to variables that are exposed to subjective biases in various stages of data collection and handling and that support the clinical objectives and collection of endpoints for the trial. (Note: The variables cannot be pharmacologic targets, such as AE/SAEs or the endpoints themselves.3,4)
Ensure adequate setup in hybrid situations: More and more pharma companies ask CROs to use their risk management tools or even to maintain their own risk management standard operating procedures (SOPs). This, however, requires detailed discussions between the sponsor and its CRO partner to ensure ICH E6 R2-compliant processes are in place. Potential questions for the CRO team to ask include:
- Is the tool/SOP ICH E6 R2 compliant, and is the CRO team trained on the sponsor tool/SOP?
- Is the tool identifying critical data and processes?
- Is the tool establishing a risk mitigation action plan, and is there a link to the functional plans that should document these mitigations?
- Is this a full cross-functional approach involving the sponsor, CRO, and other vendors?
- Will the CRO clinical trial team members see the overall output of the risk assessment? (Note: A risk identified by another party might impact the CRO.)
- Are there regular risk reviews? Are these fully cross-functional, and are outputs available for the CRO clinical trial team members?
Have the end in mind: It is important to involve the medical writer from the onset of the clinical trial to fully understand all expectations with respect to the clinical study report (CSR). According to ICH E6 R2, the overall risk management approach and a summary of the setup and management of QTLs should be included in the CSR. The collection and documentation of QTLs especially should be detailed enough to allow sufficient information to be collected so the medical writing team is able to construct the story around potential QTL excursions, actions taken, and potential impact on the interpretation of the clinical trial results.
Clinical Trial Maintenance
Integrate multiple data sources when rereviewing risk registers to drive the right downstream decisions: According to ICH E6 R2, there is a requirement to perform risk assessments on a regular basis to assess cross functionally if mitigation actions are effective and/or if new risks are emerging, such as due to a protocol amendment or significant change in how the clinical trial is executed. While reviewing the risk register it is of utmost importance to evaluate QTLs and key risk indicator trends to understand if the likelihood of a risk is increasing. It is also paramount to include the outputs of centralized monitoring activities, which use statistical methods to identify unexpected and unusual data patterns, to support operational risk detection and appropriate decision-making.
Communicate regularly internally and externally: Real-time access to the risk register via an online portal is the preferred way of communicating risk-related information to sponsors, vendors, and other stakeholders. If this technology is not available, the risk register could be an appendix to the weekly/monthly clinical trial status reports to ensure everyone has access to the most current information. Escalations, if needed, should occur according to the principles established in the communication plan between sponsor, CRO, and other vendors.
Apply lessons learned: In case of any significant quality events or QTL excursions, a comprehensive root cause analysis will need to be performed, and actions will need to be agreed upon by the clinical trial team as part of the ongoing trial management process. When such issues occur, some lessons-learned activities should take place and the clinical trial team should revisit the risk register to assess whether the risk (of the issue occurring) was not identified, or if risk mitigations were not appropriately designed or implemented.
Conclusion
Completing both a more formal risk assessment before the clinical trial activities start and regular rereviews of the risks during the life cycle of the clinical trial take time and resources, but if done properly they will pay off in the long run.
Benefits of the more formal process include but are not limited to:
- Bringing potential subject safety or data integrity risks to the surface much earlier, which will contribute to higher data quality and improved subject safety
- Prioritizing risks so they receive appropriate level of attention
- Supporting the development of effective, risk-based trial plans that focus on the critical trial elements
- Reducing the impact and probability of risks by designing appropriate mitigations prior to clinical trial start
- Reducing the number of protocol amendments
While there may be some change management challenges to overcome, the benefits clearly outweigh any perceived additional efforts.
References
- https://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E6/E6_R2__Step_4_2016_1109.pdf
- http://www.appliedclinicaltrialsonline.com/transitioning-risk-based-monitoring-approach-risk-based-quality-management
- https://www.ppdi.com/services/clinical-development/clinical-trial-monitoring/risk-management-white-paper
- https://www.clinicalleader.com/doc/taking-control-of-quality-tolerance-limits-in-clinical-trials-0001